Ошибка
Перейти на…
Перейти на…Системные требования для ЭУМКОбъявленияСПИСОК СТУДЕНТОВ, НЕ СДАВШИХ И НЕ ЯВИВШИХСЯ НА ЭКЗАМЕНРАСПИСАНИЕ ЭКЗАМЕНОВСТУДЕНТЫ, ОСВОБОЖДЕННЫЕ ОТ УСТНОЙ ЧАСТИ ЭКЗАМЕНАЭЛЕКТРОННЫЙ ЖУРНАЛТиповая программаУчебная программаТематический план Пояснительная запискаПоложение о рейтинговой оценке знанийКРИТЕРИИ ОЦЕНКИРасписание тестированияКалендарно-тематический план лекцийГрафик отработок и консультаций расписание с изм. 29.04.2021вопросы к экзаменуТема 1 Острый и хронический гломерулонефритТЕМА 2 Инфекция мочевых путей. Тубулоинтерстициальные болезни почекИнфекции мочевых путей (УСР, 2 ч.)Тест — Инфекции мочевых путейТЕМА 3 Нефротический синдром. Амилоидоз почек. Хроническая болезнь почек и хроническая почечная недостаточностьЛекция: Нефротический синдром. Амилоидоз почек. ХБП. УСР 2 ч. доц. Солодкова И.В.Лекция: Анемии (УСР 2 часа)Тест — АнемииТЕМА 4. Геморрагические диатезы. Гемолитические анемииГеморрагические диатезыТЕМА 5.

Хронический миелоидный лейкоз (ХМЛ) у детей и подростков
Существующие методы лечения ХМЛ у детей основаны на опыте лечения взрослых. Тем не менее, в последние несколько лет, особенно с появлением ИТК, растет обеспокоенность тем, что детский хронический миелоидный лейкоз имеет много уникальных особенностей и их необходимо рассматривать и исследовать отдельно.
Эта концепция подтверждается наличием нескольких геномных различий между детьми и взрослыми пациентами, некоторые из которых связаны с ответом на терапию. Дальнейшее исследование этих открытий и оценка их влияния на течение заболевания имеют большое значение для ведения детей с ХМЛ. Например, обнаружение генетических изменений, на которые можно прицельно воздействовать таргетными препаратами параллельно с применением ИТК, что в настоящее время невозможно без трансплантации костного мозга.
Существующие прогностические шкалы, позволяющие предсказывать исход, не учитывают особенностей детей. Систематическое исследование большого числа детей с ХМЛ, получавших ИТК, может помочь получить информацию для улучшения лечения.
До сих пор неизвестно, почему у подгруппы детей с ХМЛ, получавших ИТК, развиваются побочные эффекты. Вероятно, эти ответы опосредуются генетическими факторами организма. Изучение возможных факторов организма, связанных с токсичностью ИТК, может способствовать выявлению подгруппы пациентов, которым может помочь альтернативное лечение, такое как трансплантация костного мозга.
Еще одной областью клинических испытаний является вмешательство, направленное на уменьшение длительных побочных эффектов, например применение гормона роста у детей с низким ростом, обусловленным приемом ИТК. Принимая во внимание ожидаемую долгую продолжительность жизни детей и относительно короткий период — около 15 лет, — в течение которого использовался ИТК, все еще существует неопределенность относительно потенциальных побочных эффектов у пациентов, подвергавшихся воздействию ИТК в течение длительного времени.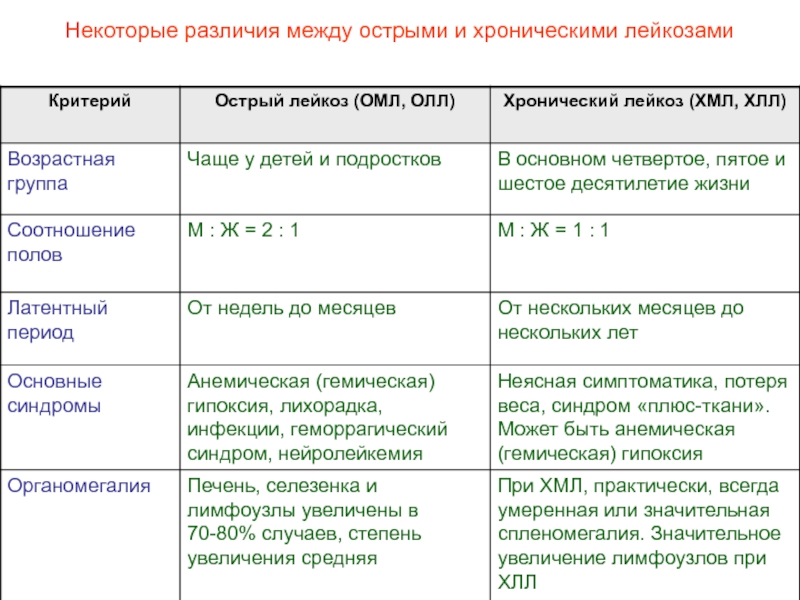
Наконец, приблизительно у 40% пациентов, которые оставались в молекулярной ремиссии в течение нескольких лет, прекращение приема ИТК было связано с безрецидивным состоянием. Областью исследования также являются попытки выявить пациентов, для которых можно безопасно прекратить применение ИТК.
Хронический миелолейкоз: лечение, симптомы, диагностика
Хронический миелолейкоз (ХМЛ, хронический миелоидный лейкоз) занимает третье место среди онкогематологических заболеваний. Еще несколько десятилетий назад болезнь плохо поддавалась лечению, но современные препараты позволяют значительно продлить жизнь пациентов и при этом сохранить ее качество.
Чаще всего миелолейкоз развивается у взрослых, примерно в 90% случаев болезнь обнаруживается именно в хронической форме. Чаще диагностируется у мужчин. Фактор, увеличивающий риск заболевания, – воздействие ионизирующего излучения.
Хронический миелоидный лейкоз относится к группе миелопролиферативных заболеваний.
Болезнь развивается из-за случайной хромосомной поломки, которая происходит в стволовой клетке. Стволовые клетки способны дифференцироваться в различные виды клеток, которые выполняют свои функции в организме, в том числе и в форменные элементы крови. Поломка получила название «филадельфийской» хромосомы. В клетках с поломанной хромосомой образуется онкобелок – тирозинкиназа, обладающий повышенной активностью и нарушающий работу нормальных клеток костного мозга. Увеличивается количество «молодых» форм лейкоцитов, которые постепенно размножаются и заполняют костный мозг, а затем выходят в периферическую кровь, проникают в печень и селезенку.
Хронический миелолейкоз не относится к наследуемым патологиям.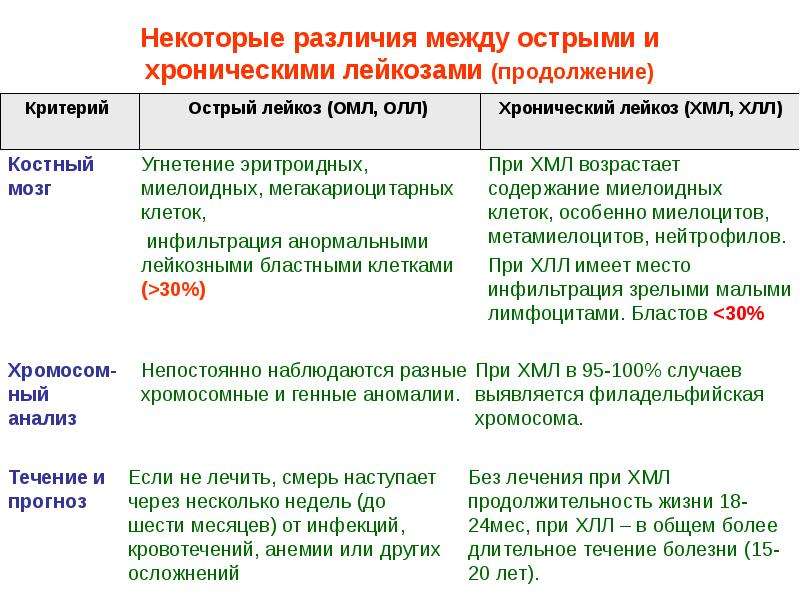
Стадии заболевания
В течении хронического миелолейкоза выделяют 4 фазы:
Хроническая фаза
Менее 10% клеток в крови и костном мозге являются патологическими. Клинические признаки в этот период отсутствуют, но могут наблюдаться слабость, утомляемость, чувство тяжести в животе. Фаза может длиться до пяти лет, поэтому при наличии данных симптомов очень важно проводить ежегодные исследования показателей крови, а при их отклонениях обязательно получить консультацию специалиста.
Фаза ускорения (акселерации)
От 10% до 19% клеток в крови и костном мозге являются патологическими. Появляются признаки анемии, из-за нехватки тромбоцитов развиваются кровотечения, повышается утомляемость и усиливается риск развития инфекционных заболеваний, возможно развитие неврологических и зрительных нарушений, может появиться боль в суставах. Пациент нуждается в госпитализации и лечении.
Пациент нуждается в госпитализации и лечении.
Бластный криз
Из-за резкого увеличения количества патологических клеток (более 20%) возникает бластный криз, который по клиническим признакам напоминает острый миелобластный лейкоз. Это быстро прогрессирующее состояние, сопровождающееся значительным повышением температуры тела, увеличением селезенки, слабостью. Бластный криз – угрожающее жизни и плохо поддающееся лечению состояние.
Фаза ремиссии
Благодаря эффективному лечению, пациенты достигают ремиссии. В этот период максимально уменьшается количество клеток с «филадельфийской» хромосомой, благодаря чему нормализуется количество лейкоцитов, гемоглобина и тромбоцитов. У пациента в этот период отсутствуют клинические симптомы болезни.
Симптомы ХМЛ
На начальном этапе симптомы заболевания отсутствуют.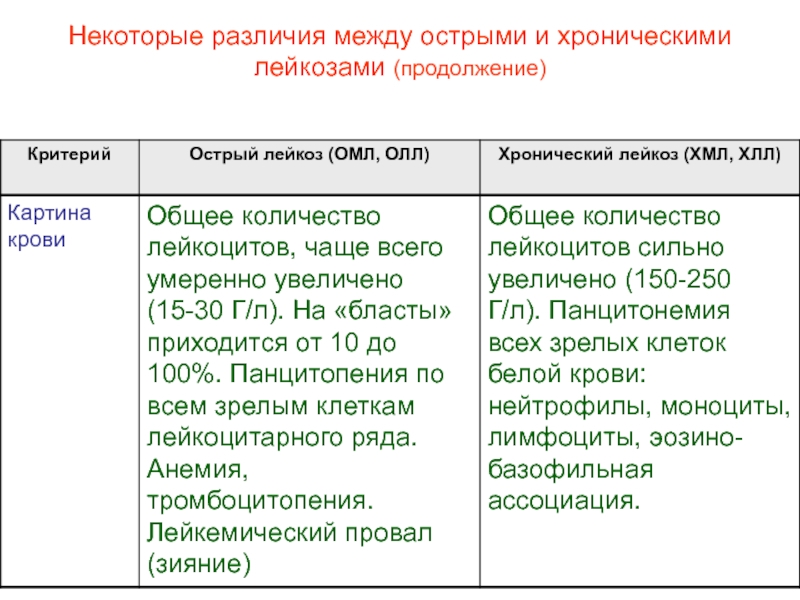
Затем возникают и постепенно усиливаются вялость, утомляемость, повышенная потливость (особенно ночью), чувство тяжести в левом боку из-за увеличения селезенки. Пациенты теряют аппетит и вес. Из-за накопления патологических клеток может появиться боль в суставах, а также — повышаться температура. В связи с тем, что в крови увеличивается количество аномальных клеток и возникает дефицит «здоровых» клеток, снижается иммунитет и усиливается предрасположенность к инфекционным заболеваниям. Развивается анемия, бледность кожных покровов, на ногах и на слизистой оболочке полости рта появляется сыпь в виде мелких красных точек (петехии), могут появляться кровоподтеки или развиваться кровотечения.
Для установления точной причины обнаруженных симптомов следует обязательно пройти полное диагностическое обследование. Обратитесь в LISOD, чтобы быть уверенным в точности результатов и в высоком качестве диагностики.
Диагностика
Онкогематолог.
 Доктор медицинских наук. Заслуженный врач Украины.
Доктор медицинских наук. Заслуженный врач Украины.
 Доктор медицинских наук. Заслуженный врач Украины.
Доктор медицинских наук. Заслуженный врач Украины.Первый шаг — выполнение анализов крови. У пациентов с ХМЛ увеличивается количество «молодых» форм лейкоцитов. Однако для постановки окончательного диагноза обязательно должна быть проведена биопсия костного мозга с последующим гистологическим исследованием.
В Больнице израильской онкологии LISOD выполняют различные виды биопсий. Все они проводятся с высококлассным анестезиологическим обеспечением, что полностью исключает болевые ощущения при проведении данной процедуры. Полученный материал направляется на исследование, позволяющее выявить «филадельфийскую» хромосому и определить количество патологических клеток. Мы можем быть уверены в правильности и точности выполнения исследований, поскольку они выполняются в референтной патологической лаборатории Германии, являющейся одной из ведущих в этом направлении.
При необходимости дообследования пациентов в LISOD есть возможность проведения всех современных диагностических исследований (КТ, ПЭТ-КТ и т. д.).
д.).
Лечение
Команда специалистов LISOD руководствуется рекомендациями международных медицинских протоколов (NCCN, AHA, EHA, ESMO). Цель лечения — максимальное снижение количества атипичных клеток в крови и костном мозге. При этом возможно достичь долговременной ремиссии.
В LISOD определением тактики лечения для каждого пациента занимается консилиум специалистов. Это дает возможность учесть все нюансы заболевания и назначить наиболее эффективное лечение с учетом возраста пациента, общего состояния здоровья, фазы заболевания.
Основные препараты для лечения ХМЛ — ингибиторы тирозинкиназы. Пациенты, обратившиеся в LISOD, имеют возможность лечения препаратами последних поколений из этой группы. При развитии бластного криза лечение проводится по схеме лечения острых лейкозов.
Лечение хронического миелолейкоза в LISOD позволяет пациентам достичь длительной ремиссии и после лечения вернуться к привычной жизни.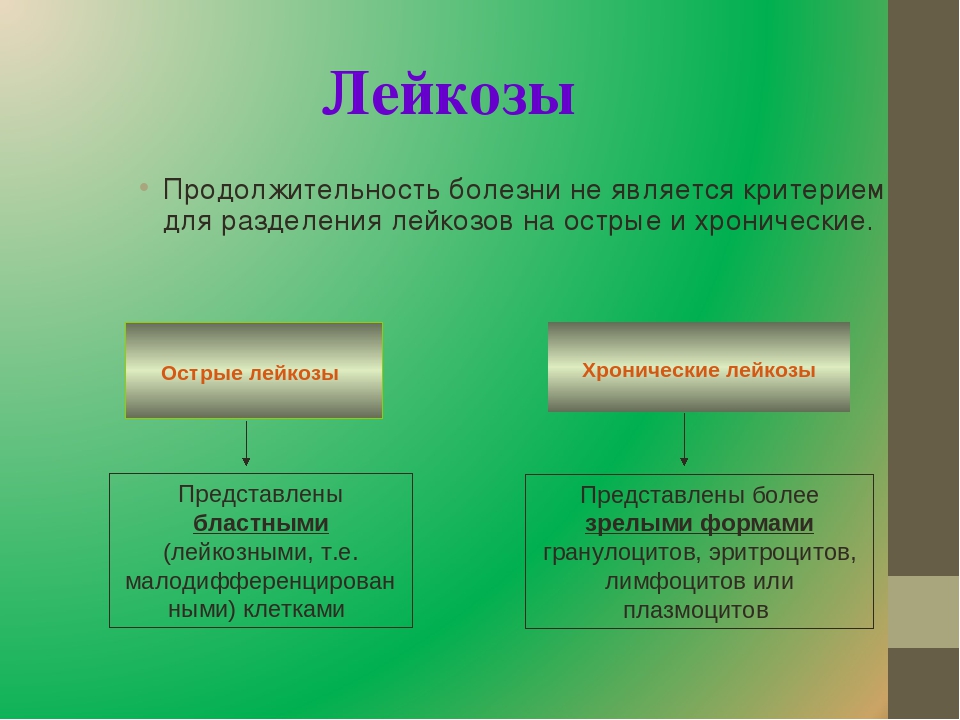 На всех этапах лечения специалисты больницы контролируют состояние здоровья пациента, при необходимости — меняют схему лечения и назначают сопроводительную терапию.
На всех этапах лечения специалисты больницы контролируют состояние здоровья пациента, при необходимости — меняют схему лечения и назначают сопроводительную терапию.
Лейкемия — Docrates
Лейкемия – это самый распространенный вид рака у детей. Она не является наследственным заболеванием, однако иногда может диагностироваться у представителей нескольких поколений одной семьи. При некоторых наследственных или врожденных заболеваниях механизм восстановления генетического материала (ДНК) нарушен, и значительно повышается риск острой формы лейкоза.
По характеру течения заболевания лейкемия делится на две основные формы: острую и хроническую.
Острая лейкемия (острый лейкоз) подразделяется на острую миелобластную лейкемию (острый миелобластный лейкоз, острый миелолейкоз, острый миелобластоз) и острую лимфобластную лейкемию. Данные формы, в свою очередь, также имеют отдельные подтипы.
Данные формы, в свою очередь, также имеют отдельные подтипы.
Говоря о хронической лейкемии, следует упомянуть две наиболее часто встречающиеся формы: хроническую лимфобластную лейкемию (самый распространенный вид лейкоза, около 150 диагностируемых случаев заболевания в год) и хроническую миелобластную лейкемию. К более редким хроническим формам лейкоза относятся: Т-клеточный пролимфоцитарный лейкоз, волосковоклеточный лейкоз и лейкоз из больших гранулярных лейкоцитов.
На сегодняшний день существуют передовые методы лечения лейкемии и найдены эффективные способы терапии различных форм этой болезни. Хроническая миелобластная лейкемия стала первым видом рака крови, для терапии которого был разработан комплекс целенаправленного лечения, благодаря чему в большинстве случаев течение лейкоза удается контролировать и добиваться ремиссии. Прогноз выздоровления пациентов с данным видом лейкемии значительно улучшился. При лечении других видов лейкемии с помощью химиотерапии и совмещенных с ней антител достигаются положительные результаты, а трансплантация стволовых клеток, взятых у здоровых людей (аллогенная пересадка стволовых клеток, пересадка костного мозга), все чаще дает положительные результаты.
Факторы риска лейкемии
Как правило, причину лейкемии установить не удается. Известно, однако, что, если у пациента был ранее диагностирован иной вид рака, то это может в некоторых случаях спровоцировать лейкемию (вторичный лейкоз). Еще одной причиной возникновения лейкемии могут стать различные генетические нарушения. При хронической миелобластной лейкемии (хроническом миелолейкозе) в самом раннем предшественнике клеток крови, стволовой клетке, вследствие замены участков 9 и 22 хромосом образуется мутантная (филадельфийская) хромосома, вызывающая рак крови. Причины возникновения других видов лейкемии на сегодняшний день изучены недостаточно. Некоторые факторы, например, ионизирующее излучение, контакт с растворителями (особенно бензолом) и другими химикатами, определенные виды химиотерапии, некоторые вирусы и редкие наследственные и врожденные заболевания повышают риск лейкемии.
Симптомы лейкемии
Симптомы лейкемии весьма различны и, как правило, появляются при острой форме лейкоза. Хроническая лейкемия может долгое время, даже в течение многих лет, протекать бессимптомно и диагностироваться только на основании рутинного анализа крови (особенно это касается хронической лимфобластной лейкемии). При остром лейкозе симптомы чаще всего возникают из-за недостатка клеток крови (при анемии, инфекциях и кровотечении), повышения вязкости лейкозных клеток или из-за нарушения работы органов, вызванного лейкозными клетками.
Хроническая лейкемия может долгое время, даже в течение многих лет, протекать бессимптомно и диагностироваться только на основании рутинного анализа крови (особенно это касается хронической лимфобластной лейкемии). При остром лейкозе симптомы чаще всего возникают из-за недостатка клеток крови (при анемии, инфекциях и кровотечении), повышения вязкости лейкозных клеток или из-за нарушения работы органов, вызванного лейкозными клетками.
Хронические формы лейкемии могут сопровождаться похожими, хотя и более размытыми, долго развивающимися симптомами. Кроме того, может наблюдаться повышение температуры тела при отсутствии инфекции, снижение веса и сильное потоотделение по ночам. В некоторых случаях симптомы могут быть вызваны увеличением лимфатических узлов или селезенки.
Чаще всего при вышеуказанных симптомах диагностируется анемия (недостаток эритроцитов), при которой уровень гемоглобина падает ниже нормы, пониженные лейкоциты (белые клетки крови) при острых формах лейкемии и повышенные лейкоциты при хронических формах, а также понижение тромбоцитов в крови. Анемия проявляется усталостью, бледностью, учащенным сердцебиением, шумом в ушах и общим недомоганием. Низкий уровень тромбоцитов вызывает склонность к кровотечениям, которая проявляется, например, в виде самопроизвольных синяков, носового кровотечения, кровоточивости десен и долгой кровоточивости ран. Снижение уровня лейкоцитов ведет к повышенной восприимчивости к инфекциям. Хотя при хронических формах лейкемии лейкоциты в крови повышаются, уровень здоровых белых кровяных клеток в костном мозге и крови понижается, что может привести к повышенной восприимчивости к инфекциям.
Анемия проявляется усталостью, бледностью, учащенным сердцебиением, шумом в ушах и общим недомоганием. Низкий уровень тромбоцитов вызывает склонность к кровотечениям, которая проявляется, например, в виде самопроизвольных синяков, носового кровотечения, кровоточивости десен и долгой кровоточивости ран. Снижение уровня лейкоцитов ведет к повышенной восприимчивости к инфекциям. Хотя при хронических формах лейкемии лейкоциты в крови повышаются, уровень здоровых белых кровяных клеток в костном мозге и крови понижается, что может привести к повышенной восприимчивости к инфекциям.
Диагностика при лейкемии
При острой форме лейкемии симптомы появляются довольно быстро, что заставляет пациента обратиться к врачу. Болезнь диагностируется с помощью лабораторных анализов на основании изменений картины крови. Часто хроническая лейкемия обнаруживается случайно при рутинном анализе крови. На основании анализа крови можно проследить повышение уровня лейкоцитов в долгосрочной динамике. В Финляндии для подтверждения диагноза «лейкемия» и определения типа лейкоза проводятся специальные исследования в гематологическом отделении университетской больницы. Делается биопсия костного мозга: специальной иглой врач делает прокол и берет несколько образцов костного мозга на анализ. Для постановки диагноза и определения общего состояния организма необходимо множество различных лабораторных исследований крови.
Делается биопсия костного мозга: специальной иглой врач делает прокол и берет несколько образцов костного мозга на анализ. Для постановки диагноза и определения общего состояния организма необходимо множество различных лабораторных исследований крови.
При острых формах лейкемии в крови и костном мозге обнаруживаются незрелые, бластные, клетки. При хронических формах лейкемии лейкозные клетки схожи со здоровыми, однако присутствуют в крови в гораздо большем количестве. Определение поверхностных маркеров лейкозных клеток дает возможность поставить точный диагноз в кратчайшие сроки. Хромосомные и генетические исследования подтверждают точность поставленного диагноза и зачастую позволяют прогнозировать течение болезни. Возможные хромосомные и генетические изменения могут учитываться также при отслеживании реакции организма пациента на лечение.
Лечение лейкемии
Лечение лейкемии осуществляется под руководством гематолога. В Финляндии пациентов с острыми формами лейкемии обычно направляют на лечение в гематологическое отделение университетской больницы. Терапия хронических форм может проводиться также в отделениях гематологии других медучреждений: лечение, как правило, проводится амбулаторно, нахождения в стационаре не требуется. Стандартное лечение лейкемии включает терапию различными химиотерапевтическими препаратами, в некоторых случаях в сочетании с антителами, распознающими раковые клетки. Наряду с этим проводится поддерживающая терапия, например, прием препаратов крови, антибиотиков, лекарств от тошноты, препаратов, защищающих слизистую желудочно-кишечного тракта и почки.
Терапия хронических форм может проводиться также в отделениях гематологии других медучреждений: лечение, как правило, проводится амбулаторно, нахождения в стационаре не требуется. Стандартное лечение лейкемии включает терапию различными химиотерапевтическими препаратами, в некоторых случаях в сочетании с антителами, распознающими раковые клетки. Наряду с этим проводится поддерживающая терапия, например, прием препаратов крови, антибиотиков, лекарств от тошноты, препаратов, защищающих слизистую желудочно-кишечного тракта и почки.
При острых формах лейкемии пациент сначала получает большую дозу химиотерапии (индукционная терапия) для того, чтобы удалить лейкозные клетки из крови костного мозга (добиться так называемой морфологической ремиссии). При положительном результате данного этапа лечения проводятся разные виды терапии для улучшения реакции организма на лечение, на медицинском языке называемые консолидационной терапией. Если этими методами вылечить лейкемию не удается, или она рецидивирует, может быть рассмотрен вариант пересадки донорских стволовых клеток (аллогенной трансплантации). Поиск подходящего донора – задача довольно сложная, так как у донора и пациента должен быть идентичный тип ткани. На роль донора чаще всего подходят родные братья или сестры пациентов, но подходящий донор может быть также найден в соответствующем реестре добровольцев. Поиск ведется по Финскому реестру доноров стволовых клеток, находящемуся в ведомстве Финского Красного Креста. В реестре насчитывается около 22 000 добровольцев. В распоряжении финских врачей есть также аналогичные зарубежные реестры, общее число потенциальных доноров в которых составляет около 22 млн. человек.
Поиск подходящего донора – задача довольно сложная, так как у донора и пациента должен быть идентичный тип ткани. На роль донора чаще всего подходят родные братья или сестры пациентов, но подходящий донор может быть также найден в соответствующем реестре добровольцев. Поиск ведется по Финскому реестру доноров стволовых клеток, находящемуся в ведомстве Финского Красного Креста. В реестре насчитывается около 22 000 добровольцев. В распоряжении финских врачей есть также аналогичные зарубежные реестры, общее число потенциальных доноров в которых составляет около 22 млн. человек.
Пересадка стволовых клеток представляет собой очень сложную процедуру, и некоторым пациентам она не может быть проведена. В некоторых случаях данная процедура может привести к смерти. Смерть может наступить из-за токсичности процедуры, возможного отторжения чужеродных клеток и рецидива болезни.
Повторная лейкемия часто встречается у взрослых пациентов. Большинство детей, перенесших лейкоз, выздоравливает.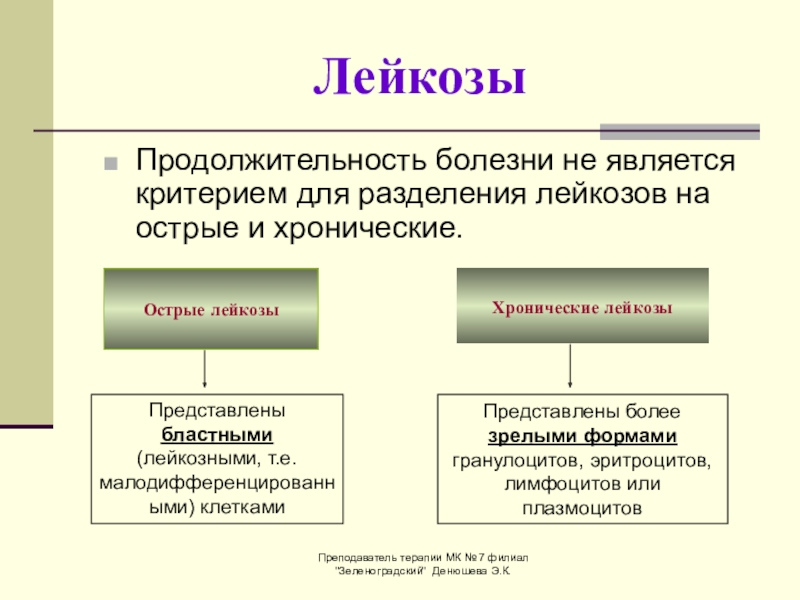 Благодаря современному лечению самую распространенную форму лейкемии у детей, острый лимфобластный лейкоз, удается вылечить в более чем 80% случаев.
Благодаря современному лечению самую распространенную форму лейкемии у детей, острый лимфобластный лейкоз, удается вылечить в более чем 80% случаев.
Лечение лейкоза (лейкемии) в Израиле: отзывы, цены, инновации.
Лейкоз (лейкемия, рак крови) — это группа онкологических заболеваний крови и костного мозга. Острые лейкозы и хронические в стадии обострения протекают стремительно и нуждаются в эффективном и оперативном лечении. Онкогематология в Израиле находится на очень высоком уровне, что позволяет успешно лечить даже такие сложные онкологические заболевания, как острый лимфобластный или миелобластный лейкоз.
Почему лечение лейкемии в Израиле столь эффективно? Направлению онкогематологии здесь уделяется колоссальное внимание: ряд клиник известны на весь мир своими отделениями по трансплантации костного мозга. Кроме этого в лечении здесь применяются новейшие протоколы и препараты лечения рака крови, которые позволяют в гораздо большем числе случаев добиваться стойкой и продолжительной ремиссии.
Типы рака крови
В отличие от большинства раковых заболеваний, лейкозы не формируют опухоль, которую можно описать и присвоить ей ту или иную стадию и в соответствии с которой будет назначаться лечение. Рак крови в целом поражает костный мозг и метастазирует в другие органы — печень, селезенку, лимфатические узлы. Выбор стратегии лечения зависит от результатов лабораторных исследований образцов опухолевых клеток, типа лейкемии, возраста пациента, его общего состояния.Глобально все лейкозы делятся на две большие группы: острые и хронические. Острый лейкоз не станет хроническим и хронический не может стать острым, за исключением хронического миелобластного лейкоза, на фоне которого может развиться острый лейкоз.
Это деление основано на зрелости клеток крови, которые мутируют и становятся раковыми. Острые лейкозы прогрессируют быстрее, хронические медленнее.
Наиболее часто среди острых лейкозов встречаются острый лимфобластный и острый миелобластный лейкозы. Каждый вид острого и хронического лейкозов имеет несколько подтипов.
Острые лейкозы связаны с утратой мутировавших клеток способности к созреванию, постепенно они замещают собой зрелые клетки и в крови начинается ощущаться нехватка зрелых клеток, что приводит к появлению клинических симптомов заболевания.
После первого основного курса лечения (химиотерапия, таргетная терапия, пересадка костного мозга) ремиссии добиваются 80-90% пациентов, однако у примерно половины случается рецидив, поэтому о полном излечении можно говорить примерно в 30-40% случаев.
Среди хронических лейкозов наиболее часто встречаются хронический лимфоцитарный лейкоз, хронический миелоцитарный лейкоз и хронический моноцитарный лейкоз.
В течении хронических лейкозов можно выделить две стадии:
1 стадия (моноклоновая): может характеризуется наличием одного клона опухолевых клеток и может протекать годами.
2 стадия (поликлоновая): характеризуется появлением множества бластов (незрелых клеток костного мозга) и называется бластный криз. Симптоматика бластного криза похожа на острые лейкозы. При бластном кризе в крови или костном мозге обнаруживается более 20 % миелобластов или лимфобластов, хлорома (опухоль), состоящая из клеток костного мозга и расположенная в любом органе за пределами костного мозга.
При бластном кризе в крови или костном мозге обнаруживается более 20 % миелобластов или лимфобластов, хлорома (опухоль), состоящая из клеток костного мозга и расположенная в любом органе за пределами костного мозга.
Пятилетняя выживаемость при хронических типах лейкоза составляет примерно 85-90%.
Преимущества лечения лейкемии в Израиле
- высокий уровень развития онкогематологии и трансплантологии в Израиле;
- мультидисциплинарный подход к лечению, когда над каждым случаем лейкоза трудится команда специалистов разных направлений;
- современное оборудование и лаборатории, позволяющие проводить успешные пересадки костного мозга.
Инновационные методы лечения лейкемии в Израиле
- лечение современными таргетными препаратами на основе моноклональных антител, позволяющими добиваться хороших результатов в лечении с минимальными побочными эффектами;
- иммунотерапия новейшим методом CAR-T с использованием собственных “перепрограммированных” лимфоцитов;
- новые методы и препараты химиотерапии острых и хронических лейкозов.

Методы лечения лейкоза
Стратегия лечения зависит от типа лейкоза. Острые лейкозы нуждаются в немедленной и агрессивной терапии, хронические могут длительно протекать без лечения, зачастую больной может всю жизнь прожить со своим заболеванием.Химиотерапия
Это основной метод лечения рака крови. Химиотерапия острых лейкозов проводится в три этапа:индукция: короткий и интенсивный курс, который длится около 3-4 недель. Это лечение должно убить основную массу раковых клеток, часто после этого этапа наступает полная ремиссия. На этом лечение не заканчивается, поскольку без последующей химиотерапии рак часто рецидивирует.
консолидация: также интенсивный курс, который проводится в течение нескольких месяцев с перерывами. Призван поддержать лечение 1 этапа и закрепить его.
пост-консолидация (поддерживающая химиотерапия): менее интенсивный, но длительный прием препаратов — около 2-3 лет.
Обычно назначается комбинация нескольких препаратов, в течение курса лечения препараты могут заменяться. Такая схема лечения должна вызвать длительную ремиссию или навсегда победить рак крови.
Такая схема лечения должна вызвать длительную ремиссию или навсегда победить рак крови.
Для лечения хронических лейкозов химиотерапия проводится при ухудшении состояния пациента. Длительность терапии и препараты назначаются индивидуально. После активного курса лечения и стабилизации состояния может быть назначена пожизненная терапия химиопрепаратами.
Химиотерапия может вводиться традиционно — методом внутривенных вливаний или интратекально — непосредственно в спинной мозг. Интратекальная химиотерапия может назначаться при лечении острого лимфобластного лейкоза у взрослых, когда имеет место тенденция к распространению процесса в головной мозг. Проводится она в сочетании с обычной, классической химиотерапией.
Пересадка костного мозга
Если обычная химиотерапия не дала результата, пациенту может быть предложена трансплантация костного мозга.Это позволяет врачам использовать более высокие дозы химиотерапии (иногда в комплексе с радиотерапией) , чтобы убить раковые клетки.
 Высокие дозы препаратов полностью убивают костный мозг, включая здоровые клетки. После первого этапа пациенту проводится пересадка стволовых клеток для восстановления здорового костного мозга.
Высокие дозы препаратов полностью убивают костный мозг, включая здоровые клетки. После первого этапа пациенту проводится пересадка стволовых клеток для восстановления здорового костного мозга.Существует 2 способа трансплантации стволовых клеток в зависимости от источника замещающих стволовых клеток крови: аллогенный и аутологичный. При аллогенном методе используются донорские стволовые клетки, при аутологичном — собственные стволовые клетки пациента. Их забор производится до начала курса высокодозной химиотерапии.
Таргетная терапия
Таргетная терапия была разработана для борьбы со специфическими белками и генами раковых клеток без вреда для здоровых клеток. Таргетная терапия является одним из основных видов лечения как острых, так и хронических лейкозов и назначается после химиотерапии или в комплексе с ней.У каждого четвертого больного лимфобластным лейкозом опухолевые клетки имеют филадельфийскую хромосому. Это аномальная хромосома, образованная путем замены генетического материала между хромосомами 9 и 22, который создает новый ген BCR-ABL. Клетки с геном BCR-ABL вызывают образование белка, который обеспечивает аномальный рост клеток. Для борьбы с этим специфическим белком разработана группа препаратов, которые называются ингибиторы тирозинкиназы. Они подавляют образование этого белка, избирательно связываются с колониями клеток, содержащих его, и вызывают гибель этих клеток.
Клетки с геном BCR-ABL вызывают образование белка, который обеспечивает аномальный рост клеток. Для борьбы с этим специфическим белком разработана группа препаратов, которые называются ингибиторы тирозинкиназы. Они подавляют образование этого белка, избирательно связываются с колониями клеток, содержащих его, и вызывают гибель этих клеток.
Для таргетной терапии острого миелобластного лейкоза используются препараты, блокирующие аномальные белки FLT3, IDh2, IDh3, которые вырабатывают соответствующие гены.
Другим вариантом таргетной терапии острого миелобластного лейкоза является лечение препаратами на основе моноклональных антител (искусственного иммунного белка). Антитело прикрепляется к определенному белку CD33 на поверхности раковой клетки и действует как мишень. Препарат атакует только эти клетки и уничтожает их.
Для лечения хронических лейкозов таргетная терапия является основным методом лечения. Она показывает хорошие результаты даже у пациентов с неблагоприятными прогнозами.
Включение данной терапии в протокол лечения позволяет быстрее добиться ремиссии и сделать ее более стойкой. Для того, чтобы правильно подобрать препарат, должно быть проведено тщательное исследование биоптата костного мозга с целью выявления типа мутации раковых клеток конкретного человека.
Иммунотерапия
Новым методом в лечении лейкозов является адаптивная клеточная терапия CAR-T клетками. Из крови пациента производится забор лимфоцитов, в течение следующих двух недель специалисты искусственно размножают клетки в лаборатории и продуцируют в них антитела для борьбы с лейкемией. Для этого в них искусственным путем вводится химерный рецептор CAR (Chimeric Antigen Receptor), запускающий специфические онкомаркеры и разрушающий ткани опухоли. Пациент в это время проходит химиотерапию, направленную на угнетение собственного иммунитета. После этого пациенту вводят обратно преобразованные лимфоциты.При лечении рака крови с высокой степенью распространения опухолевых клеток в костном мозге возможно снижение эффективности CAR-T терапии.
 В таких случаях клеточная терапия дополняется химиотерапией. Кроме того, в течение первых двух месяцев после введения клеток с химерным рецептором, пациентам рекомендована трансплантация костного мозга.
В таких случаях клеточная терапия дополняется химиотерапией. Кроме того, в течение первых двух месяцев после введения клеток с химерным рецептором, пациентам рекомендована трансплантация костного мозга.Лучевая терапия
Лучевая терапия не является основным методом в лечении лейкозов, поскольку опухолевые клетки рассеяны по всему организму. Но она может назначаться в комплексе с химиотерапией, а также для уменьшения тяжести некоторых симптомов лейкозов, например, увеличенной селезенки, лимфоузлов или боли в костях. При образовании миелоидной саркомы (опухолевый очаг за пределами костного мозга) лучевая терапия может также назначаться в комплексе с химиотерапией или если химиотерапия не дала результатов. Для лечения лейкозов назначается стереотаксическая лучевая терапия, при которой используется 3D-изображение селезенки или опухоли в кости, полученное в результате КТ или МРТ, для более точного направления излучения.Кроме этого, лучевая терапия назначается перед пересадкой костного мозга для облучения всего тела.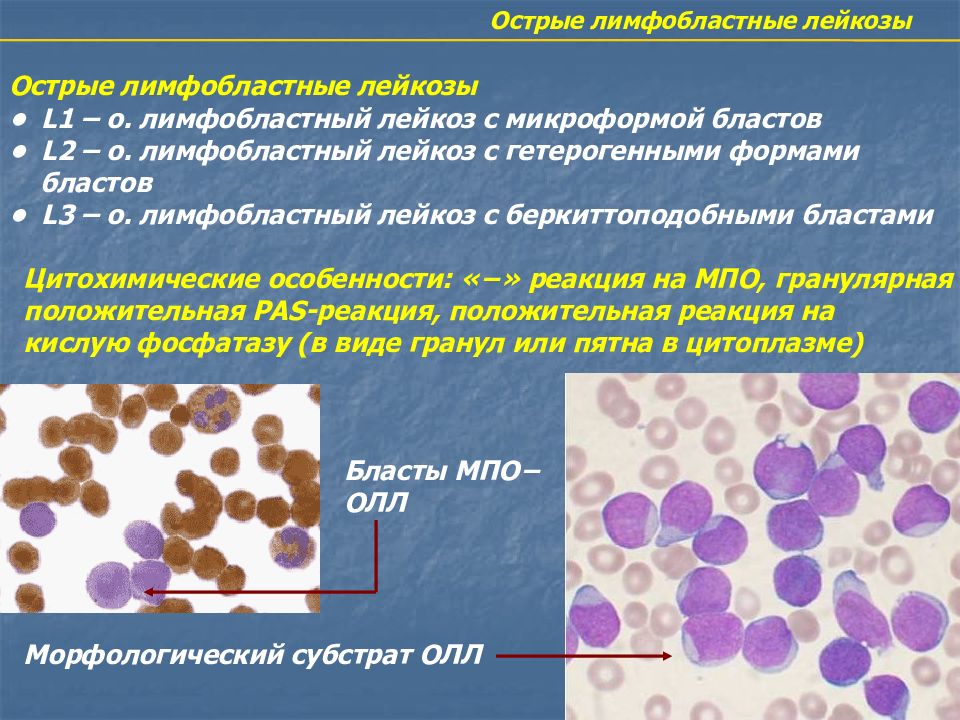 Производится это для того, чтобы максимально ослабить собственный иммунитет, чтобы пересаженный костный мозг не был отторгнут.
Производится это для того, чтобы максимально ослабить собственный иммунитет, чтобы пересаженный костный мозг не был отторгнут.
Хирургическое лечение
Хирургическое лечение практически не применяется при лейкозах, поскольку опухоль не имеет четкой локализации.В редких случаях операция может понадобиться для удаления миелоидной саркомы, гранулоцитарной саркомы или хлоромы — опухолей, которые образованы клетками костного мозга, которые вышли за его пределы и образовали очаг в любом другом органе человека.
В лечении хронических лейкозов может понадобиться такое хирургическое вмешательство, как спленэктомия — удаление селезенки, поскольку при хронических лейкозах увеличение селезенки является очень частым симптомом. Сначала увеличенную селезенку пробуют уменьшить химиотерапией и лучевой терапией, если это не помогает, то прибегают к оперативному вмешательству, так как она давит на соседние органы.
Как проходит лечение рака крови в Израиле
Этап 1. Диагностика лейкозов
Ориентировочная длительность диагностики — 2-4 дняПо прибытию в Израиль вам будет предложено пройти предварительное обследование, которое должно подтвердить поставленный ранее диагноз.
 Это будут развернутые анализы крови и ревизия биопсии, если у вас с собой имеются стекла и блоки с биопсией, взятой ранее по месту жительства. После этого состоится консультация с выбранным вами специалистом, который проведет физический осмотр, пальпацию лимфоузлов, оценит общее состояние.
Это будут развернутые анализы крови и ревизия биопсии, если у вас с собой имеются стекла и блоки с биопсией, взятой ранее по месту жительства. После этого состоится консультация с выбранным вами специалистом, который проведет физический осмотр, пальпацию лимфоузлов, оценит общее состояние.Если биопсия не проводилась ранее или ее результаты не информативны для точной постановки диагноза, ее могут провести повторно. При подозрении на лейкемию проводится биопсия костного мозга методом аспирации — с помощью специального шприца. Материал биопсии подвергается нескольким сложным анализам, в результате чего уточняется диагноз: тип лейкемии, степень агрессивности клеток, прогнозирование ответа на лечение и т.д. Именно это исследование влияет на выбор протокола лечения, поэтому оно должно быть выполнено предельно качественно.
Объем распространения опухолевых клеток и поражение других частей тела диагностируют с помощью визуализирующих исследований — КТ, ПЭТ КТ, МРТ, для обследования селезенки проводится УЗИ.
Когда проведены все исследования, лечащий врач составляет протокол лечения, ориентированный на индивидуальные особенности протекания заболевания.
Этап 2. Индивидуальные протоколы лечения лейкозов
Ориентировочная длительность: от 1 месяцаЛечение острых типов лейкозов длится несколько лет. Поскольку заболевание довольно быстро прогрессирует, лечение лучше начинать как можно раньше сразу после постановки диагноза. Первые несколько недель химиотерапия особенно интенсивна, поэтому лучше пройти в Израиле под наблюдением вашего лечащего врача. В дальнейшем вы можете продолжить курс у себя дома с соблюдением рекомендаций израильского онкогематолога.
Тактика лечения хронических типов лейкоза иная. По сути это активное наблюдение и поддерживающая терапия, сохраняющая болезнь под контролем. К активному лечению химиотерапией или таргетной терапией переходят, если болезнь обостряется и переходит в бластный криз.
При хронических лейкозах хорошие результаты дает таргетная терапия и аллогенная (донорская) трансплантация костного мозга от родственного или неродственного донора, такая тактика дает 60% случаев полной ремиссии в течение 5 и более лет.
Также в комплексе с химиотерапией может быть назначена лучевая терапия лимфоузлов, селезенки, если это не помогает, проводится спленэктомия (удаление селезенки).
Этап 3. Последующее наблюдение и профилактика рецидивов лейкемии.
Поскольку лечение острой лейкемии занимает довольно длительный период времени — 2-3 года, регулярное наблюдение является основой успешной терапии. После полного окончания лечения, если болезнь перешла в стадию ремиссии, важно продолжать наблюдение и регулярно обследоваться. Рецидивы лейкемии случаются еще в процессе лечения (последнего 1 этапа химиотерапии) или сразу после ее окончания. Если в течении 5 лет после окончания лечения рецидива не было, его вероятность в последующие годы снижается.Хронический лейкоз практически не излечим, но жить с ним можно годами, активное лечение начинается только в фазу обострения. Чтобы контролировать наступление этой фазы, нужно также проходить регулярные обследования.
Это различные анализы крови, исследования образцов костного мозга и визуализирующие исследования — ПЭТ КТ, МРТ, УЗИ.
Лейкемия даже в стадии хронического протекания негативно воздействует на иммунную систему человека, лечение химио- и таргетными препаратами также угнетает ее, поэтому у больных лейкемией повышен риск заражения различными инфекционными и бактериальными заболеваниями. После лечения лейкоза нужно стараться избегать возможного заражения даже самыми простыми инфекционными заболеваниями.
В стадии ремиссии при появлении какого-либо дискомфорта нужно немедленно обратиться к врачу и пройти обследование на предмет исключения рецидива лейкоза.
Стоимость лечения лейкозов в Израиле
Финальная стоимость диагностики и лечения лейкемии зависит от набора процедур, квалификации врача, клиники, категории палаты и т.д. Ниже приведены цены на основные процедуры, назначаемые при лейкемии.| Консультация онкогематолога: изучение предоставленной медицинской документации, составление протокола лечения. | по запросу |
| Биопсия костного мозга | по запросу |
| Ревизия биопсии. При наличии блоков и стекол с биопсией, взятой ранее. Готовность результатов в течение 72 часов. | 480-590 |
| УЗИ селезенки | по запросу |
| ПЭТ КТ – проводится при подозрении на метастазы. | 1285-1580 |
| Анализ крови включая тест на онкомаркеры. | от 189 |
| Химиотерапия один день (проведение в амбулаторном стационаре) | от 960 |
| Лекарственные препараты для биологического лечения | от 900 |
| Лучевая терапия. Стоимость 1 поля облучения | от 170 |
Получите персональную программу лечения лейкемии в Израиле
Свяжитесь с нашей медицинской группой и мы предоставим вам понятный и подробный план действий: врач-эксперт ознакомится с вашей историей болезни, на основании чего будет составлен график анализов и тестов. Вы также получите досье врачей, специализирующихся на заболевании системы крови, актуальные цены на все процедуры. Вся информация предоставляется бесплатно.
Важно помнить, что онкология — это не приговор, а этап вашего жизненного пути, который нужно преодолеть. Мы помогли многим людям и сможем помочь вам!
Звоните +972 (77) 4450480 | 8 (800) 707-6168 (Звонок из России бесплатно)
ЗАПРОСИТЬ СМЕТУ ЛЕЧЕНИЯ
Как распознать лейкоз по анализу крови? — Фонд борьбы с лейкемией
Врач-гематолог, доктор медицинских наук Сергей Семочкин про острый лимфобластный лейкоз
Врач-гематолог, профессор кафедры онкологии, гематологии и лучевой терапии РНИМУ им. Н. И. Пирогова Минздрава России, доктор медицинских наук Сергей Семочкин рассказал: можно ли распознать острый лимфобластный лейкоз (ОЛЛ) на ранней стадии и поставить диагноз по анализу крови; объяснил, как лечат ОЛЛ и кому показана трансплантация костного мозга (ТКМ).
Каковы ранние симптомы острого лимфобластного лейкоза? Можно ли их увидеть и распознать ОЛЛ?
В данном случае все достаточно просто, потому что слово «острый» означает, что заболевание внезапное и зачастую симптомы очень выразительные. Самый частый симптом — это лихорадка, т.е. повышение температуры тела. Лихорадка может быть как субфебрильной, так и ярко выраженной, до 39 градусов. Появятся изменения, связанные с поражением костного мозга. Снижение гемоглобина приведет к слабости и быстрой утомляемости. Могут увеличиться лимфатические узлы, появиться дискомфорт в брюшной полости за счет того, что увеличиваются размеры печени и селезенки. Могут быть проявления кровоточивости — даже во время чистки зубов. У некоторых пациентов ОЛЛ может начаться с неврологических проявлений – головных болей, головокружения и прочих проблем. Симптоматика обширная, но в данном случае она является достаточно острой, внезапно возникшей.
Смотрите видео на нашем сайте.
Можно ли поставить диагноз по анализу крови? Что он покажет?
Как правило, в анализе крови есть ярко выраженные показатели: изменены ростки кроветворения, количество лейкоцитов выходит за пределы нормы — может упасть ниже нормальных значений, а может стать запредельно огромным. Мне встречались пациенты, у которых количество лейкоцитов при норме от 4 до 9 тысяч повышалось до 200 тысяч на мкл. Тромбоциты тоже в ряде случаев очень сильно снижены, но главное – изменение количества лейкоцитов. Очень важным маркером является выход опухолевых клеток в кровь, когда в крови появляются незрелые ранние клетки, которые называют бластными. Если в анализе крови выявили бластные клетки, то это, скорее всего, либо острый лейкоз, либо миелодиспластический синдром.
Как пациент попадает к гематологу?
Анализ крови с характерными изменениями — повод для немедленного вызова скорой помощи и госпитализации пациента в профильный стационар. При лечении детей и подростков у онкогематолога, как правило, есть один-два дня ни диагностику, лечение необходимо начинать, как можно раньше. В диагностику входит повторный анализ крови, затем – верификация диагноза, для которой проводят биопсию костного мозга. У маленьких детей ее проводят под общей анестезией, у взрослых — под местной. С помощь небольшой иглы делаю прокол грудины или подвздошной кости. У детей пункцию грудины не делают. Полученный образец костного мозга, который выглядит как обычная пробирка с кровью, отправят в лабораторию, где для подтверждения диагноза проведут целый спектр исследований. Главный критерий – увеличение количества бластных клеток. Только по внешнему виду и по количеству бластных клеток определить вариант лейкоза – невозможно. Еще в 1913 году установили, что есть лимфоидный, а есть миелоидный вариант лейкоза. Для верификации применяются специальные лабораторные методы: иммунологические и химические. Существует специальный прибор – проточный цитометр, с помощью которого определяют маркеры, характеризующие данный тип клеток. Для определения подвида острого лейкоза, применяют целый спектр генетических исследований, чтобы выйти на более целевую терапию у этих пациентов.
Каковы причины возникновения ОЛЛ? Существует мнение, что этот вид лейкоза очень сильно взаимосвязан с экологическими проблемами, передается по наследству и часто возникает у тех, кто уже переболел каким-то онкологическим заболеванием. Правда это или нет?
Истинную причину возникновения лейкоза у взрослых можно выявить только в 5% случаев, в 95% совершенно непонятно, что там к чему привело. У детей все несколько интересней.
Как возникает лейкоз? В генетическом материале клетки возникает некая первичная мутация, которая сама по себе далеко не всегда приводит к лейкозу. В дальнейшем к этой мутации присоединяются другие, и когда болезнь все же возникает, в клетке накоплено уже много молекулярных событий, сочетание которых привело возникновению заболевания. Пик острого лимфобластного лейкоза приходится на детей от двух до четырех лет, потом заболеваемость падает. Следующий пик приходится на 18-29 лет, потом снова спад. После 60 лет — опять небольшой рост.
У части детей раннего возраста прослеживается некая врожденная составляющая этой проблемы. Встречаются случаи ОЛЛ у плода или новорожденного, когда ребенок рождается уже с заболеванием, либо заболевает в течение первого года жизни. Исследования пуповинной крови показали, что у новорожденных встречаются лейкемические поломки, врожденные мутации, которые могут привести к возникновению лейкоза. И мутацию эту вызывает наследственный фактор, сработавший во время внутриутробного развития. По разным данным, общее количество таких младенцев составляет от 1 до 5%. Дальше многое зависит от инфекционной обстановки, сложившейся вокруг ребенка. Многочисленные инфекции, перенесенные в детстве, способствуют формированию нормальной иммунной системы, которая нейтрализует наследственный фактор.
Если говорить про экологические проблемы, то с ними четкой связи не выявлено.
Влияет ли УФ-излучение, СВЧ, солнечные лучи, радиация?
В Хиросиме и Нагасаки повышенная заболеваемость держалась около 12 лет. После Чернобыля у многих пострадала щитовидная железа, но заболеваемость лейкозами не выросла. Все зависит от типа изотопов попавших в окружающую среду. В Фукусиме тоже этого не случилось, потому что концентрация радиоактивных веществ сильно разбавилась морской водой.
Вред ультрафиолета научно доказан только в отношении меланомы. Четкой связи с ОЛЛ нет. Своим бывшим пациентам мы не разрешаем посещать солярий и не рекомендуем загорать, потому что хотя связь и не доказана, совсем исключать этот фактор тоже нельзя.
Если говорить об СВЧ-излучении, домашние микроволновые печи абсолютно безопасны.
Как лечат ОЛЛ? Что ждет пациента?
Концепция лечения ОЛЛ, которая до сих поре лежит в основе протоколов лечения ОЛЛ, была разработана американским педиатром Дональдом Пинкелем еще в 1962 году. Она включает в себя четыре этапа: индукция ремиссии, консолидация, воздействие на центральную нервную систему и длительный этап поддерживающей терапии на протяжении двух-трех лет. Во всем мире проводится лечение по клиническим протоколам, разработанным в результате кооперированных исследований. Согласно некоторым работам, строгое следование протоколам повышает выживаемость пациентов на 15-20% по сравнению с индивидуализированным лечением. В протоколе прописаны все действия: от первого дня до последнего. В нем есть указания, как и в какой момент оценивать возникающие осложнение и что с ними делать. В России два центра, активно ведущих такие протоколы. Центр им. Дмитрия Рогачева, где Александр Исаакович Карачунский в течение многих лет, с начала 1990-х, ведет серию протоколов «Москва — Берлин». Каждые пять лет дизайн протоколов пересматривают, чтобы улучшить лечение отдельных категорий пациентов. уже в течение многих лет с начала 90-х годов серию протоколов Москва-Берлин. Каждые пять лет меняется дизайн протоколов, направленных на улучшение лечения отдельных категорий пациентов. Во взрослой практике — это НМИЦ гематологии, где ведут кооперированные исследования по острому лимфобластному лейкозу у взрослых.
В каких случаях показана трансплантация костного мозга (ТКМ)?
В отличие от острого миелоидного лейкоза, показаний к аллогенной (от донора) ТКМ меньше. Ее назначают пациентам, которые не достигли ремиссии в указанные протоколом сроки или имеют неблагоприятный цитогенетический вариант заболевания. В детской практике выздоравливают более 90% детей, и примерно 15-20% являются кандидатами для аллогенной ТКМ. У взрослых процент пациентов нуждающихся в трансплантации несколько выше, за счет того, что генетических операций высокого риска становится намного больше и ответ на стандартное лечение хуже. Когда мы обсуждали хронический миелолейкоз, там фигурировала филадельфийская хромосома — транслокация (9;22). При ОЛЛ это абсолютно негативный фактор прогноза. У детей такая мутация встречается меньше чем в 5% случаев, у людей старше 50-60 лет примерно половина В-линейных ОЛЛ будет с филадельфийской хромосомой. В отличие от хронического миелолейкоза, применение ингибиторов тирозинкиназы при остром лимфобластном лейкозе не столь успешно. Вот поэтому во взрослой практике ТКМ необходимо проводить примерно 30% пациентов. Возрастной порог для аллогенной ТКМ — в районе 55 лет, это разумно.
Как часто случаются рецидивы с ОЛЛ?
Если мы говорим про взрослых людей, то рецидивы случаются почти в 40% случаев. Бывают ранние рецидивы, которые случаются прямо на терапии. В таком случае необходимо менять лечение, делать его более интенсивным и тяжелым. В таких случаях, как правило, показана ТКМ. Поздний рецидив может случиться и через 20 лет. К сожалению мы не можем убрать причину, которая вызывает это заболевание — оно может вернуться.
Можно ли планировать беременность после ОЛЛ?
Длительная химиотерапия нарушает фертильность, поэтому лучше провести криоконсервацию спермы/яйцеклетки, а еще лучше эмбриона — это более надежный способ. У мужчин, как правило, серьезно нарушается сперматогенез, но у женщин дело обстоит несколько лучше. Вероятность забеременеть и выносить здорового ребенка высока. Если прошло не менее пяти лет в ремиссии, никаких ограничений нет.
Может ли беременность быть провоцирующим фактором для рецидива?
Скорее, нет. Это не такое частое явление, как при некоторых других заболеваниях, где беременность действительно может стать провоцирующим фактором.
Передается ли ОЛЛ по наследству?
Лимфобластный лейкоз – редкое заболевание, поэтому вероятность того, что он случится у ребенка, рожденного от родителей после ОЛЛ, крайне мала.
Как будут лечить ОЛЛ в будущем?
Представляется, что в основе лечения онкологических заболеваний в будущем станет активация собственного иммунитета. Нам необходимо настроить иммунную систему таким образом, чтобы она распознавала и убирала раковые клетки. Сейчас мы находимся на раннем этапе развития CAR-T-терапии, но через какое-то время технологии настолько усовершенствуются, что, скорее всего, она станет одним из основных методов терапии при целом ряде онкогематологических заболеваний. Суть метода заключается в том, что у пациента собирают его собственные Т-лимфоциты и отправляют в специальную лабораторию. Эта лаборатория может быть в другом городе, стране — не важно. В лаборатории эти Т-лимфоциты перепрограммируются: в них появляется информация об опухолевых клетках, присутствующих в организме пациента. После перепрограммирования Т-лимфоциты вводят обратно пациенту, она находят раковые клетки и возникает ремиссия. Основные проблемы – создать качественный процесс распознавания и разработать стандартные протоколы лечения.
Много вопросов возникает в понимания биологии заболевания, потому как каждый конкретный случай весьма индивидуален. Мы знакомы только с грубыми поломками, но каждая отдельная поломка провоцирует различное течение болезни. Мы уже сейчас можем полностью секвенировать геном опухолевой клетки и главное научиться понимать, что в патогенезе является ключевым и как на это можно воздействовать, тогда мы ближе подойдем к полному излечению болезни. За этим будущее.
Хронические миелолейкозы | Компендиум
Заболевания, относящиеся к группе хронических миелопролиферативных заболеваний (ХМПЗ), возникают в результате злокачественной трансформации полипотентной гемопоэтической стволовой клетки костного мозга и последующей клональной пролиферации клеток одной или более линий миелопоэза, сохраняющих способность к дифференцировке [1–3]. Различные формы ХМПЗ имеют ряд сходных и перекрывающихся морфологических и клинико-гематологических признаков (спленомегалия, лейкоцитоз, тромбоцитоз, увеличение количества мегакариоцитов и развитие фиброза в костном мозге). Возникновение фиброза, а в ряде случаев и явлений склероза, носит реактивный характер. Обусловлено оно пролиферацией фибробластов, являющихся основными клеточными элементами кроветворного микроокружения костного мозга, не относящихся к клону злокачественных клеток.
В то же время имеются и существенные различия клинико-лабораторных данных, на которых основывается современная классификация ХМПЗ [4–6]. При изучении мазков периферической крови и костного мозга, гистологическом исследовании трепанобиоптатов, применении цитогенетических и молекулярно-биологических методов удается выделить следующие основные формы ХМПЗ (табл. 21).
Хронический миелолейкоз
Хронический миелолейкоз (ХМЛ) является клональным злокачественным процессом. У 95% больных в клетках костного мозга и периферической крови определяется так называемая филадельфийская (Ph’) хромосома, возникающая в результате транслокации генетического материала между хромосомами 9 и 22 — t (9; 22) (q34.1; q11.21). У 5% больных с ХМЛ Ph’ хромосома не определяется (так называемые Ph’ негативные случаи). Но при этом у некоторых больных выявляются характерные аномалии BCR-ABL [8].
На долю ХМЛ приходится около 15–20% всех случаев лейкозов у взрослых и 5% у детей. В США ежегодно регистрируется 3400 новых случаев заболевания ХМЛ [2]. Возрастной пик заболеваемости приходится на 4–5-е десятилетие жизни. Среди больных несколько преобладают лица мужского пола.
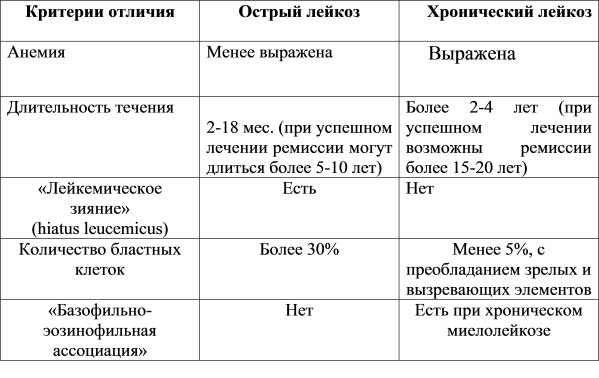
В гематологическом плане заболевание характеризуется выраженным лейкоцитозом, сочетающимся с базофилией и эозинофилией. Лейкоцитоз обусловлен увеличением в периферической крови количества зрелых и незрелых нейтрофилов [2, 3, 6].
Хроническая фаза заболевания
Общее количество лейкоцитов в периферической крови колеблется в широких пределах, но обычно выше 50•109/л. У 70–90% больных количество лейкоцитов превышает 100•109/л, а у 25% — выше 350•109/л [1–4, 6]. Выраженный лейкоцитоз часто наблюдается при ХМЛ у детей. В хронической фазе заболевания содержание миелобластов обычно колеблется в пределах 2–3%, а общее количество промиелоцитов и миелобластов не превышает 15–20% от общего количества лейкоцитов в периферической крови или костном мозге. Количество незрелых клеточных элементов гранулоцитарного ряда (промиелоцитов и миелоцитов) увеличивается по мере прогрессирования процесса при одновременном уменьшении процентного содержания палочкоядерных и сегментоядерных лейкоцитов. В нейтрофилах периферической крови и костного мозга в хронической фазе ХМЛ, как правило, не наблюдается диспластических изменений. У некоторых больных отмечаются выраженная эозинофилия и базофилия (так называемая эозинофило-базофильная ассоциация) и наличие многочисленных незрелых клеток эозинофильного и базофильного ряда. У большинства больных с ХМЛ определяется также абсолютный моноцитоз. Выраженный моноцитоз в ранней фазе заболевания позволяет предположить наличие хронического миеломоноцитарного лейкоза (ХММЛ). Во время установления диагноза у многих больных с ХМЛ определяется нормоцитарная нормохромная анемия, признаки анизоцитоза и пойкилоцитоза. Анемия прогрессирует по мере увеличения количества лейкоцитов в периферической крови. Одновременно в крови определяется небольшое количество ядросодержащих клеточных элементов эритробластического ряда. Почти у 50% больных с ХМЛ отмечается тромбоцитоз. Количество тромбоцитов в крови увеличивается по мере развития заболевания, нередки случаи, когда тромбоцитоз составляет 1000•109/л. В мазках периферической крови больных обнаруживаются гигантские формы тромбоцитов, пластинки с аномальной грануляцией, ядра мегакариоцитов.
В периферической крови больных нередко еще за несколько месяцев до манифестации ХМЛ определяются псевдопельгеровские лейкоциты. Помимо ХМЛ, эта приобретенная аномалия гранулоцитов может обнаруживаться у больных с идиопатическим миелофиброзом, при ОМЛ, иногда при неходжкинских злокачественных лимфомах, инфекционных процессах, действии ряда токсических веществ.
В костном мозге больных с ХМЛ обнаруживается гиперклеточность, обусловленная преобладанием нейтрофилов и незрелых клеток гранулоцитарного ряда. Резко увеличено соотношение клеток гранулоцитарного и эритробластического ряда, составляющее нередко 20:1 вместо 3:1 в норме. В костном мозге определяется тот же спектр клеток гранулоцитарного ряда, что и в периферической крови, но с бо’льшим преобладанием незрелых клеток (промиелоцитов и миелоцитов). Отмечается преимущественно нормобластический эритропоэз, но у отдельных больных в хронической фазе заболевания — мегалобластический и признаки дисэритропоэза. Количество мегакариоцитов увеличено, как и при многих других миелопролиферативных заболеваниях. Палочки Ауэра в цитоплазме клеток, в отличие от ОМЛ, определяются редко. Их появление может служить предвестником развития бластного криза.
Приблизительно у 30% больных ХМЛ в костном мозге и селезенке определяются крупные клетки с пенистой цитоплазмой и эксцентрично расположенным ядром, напоминающие клетки, обнаруживаемые при болезни Гоше. Эти псевдо-Гоше-клетки изредка встречаются также при врожденной дисэритропоэтической анемии, множественной миеломе, иммунобластной лимфоме, лимфогранулематозе. Считается, что их появление обусловлено неспособностью клеточных гликоцереброзидаз расщеплять гликоцереброзиды, образующиеся в больших количествах при усилении распада лейкоцитов. Подобно истинным клеткам Гоше они PAS-положительны, окрашиваются масляным красным О, СЧВ, обладают активностью КФ [2, 3, 5].
В костном мозге больных ХМЛ при окраске по Романовскому—Гимзе встречаются также крупные, так называемые цвета голубого моря гистиоциты. Эти клетки с яркой сине-зеленой цитоплазмой, как и псевдо-Гоше-клетки, дающие положительную реакцию на гликоген и липиды, изредка выявляются при идиопатической тромбоцитопенической пурпуре, ОМЛ, ОЛЛ, неходжкинских злокачественных лимфомах. Возможно, что оба типа клеток в действительности представляют две разные стадии развития макрофагов, содержащих остатки фагоцитированных нейтрофилов [1].
Получение и исследование пунктатов костного мозга, как и периферической крови, не только обязательно для установления диагноза ХМЛ. Оно позволяет провести цитогенетические исследования, определить реарранжировку генов BCR и ABL, изучить характер роста клеток-предшественников в культуре in vitro [6–8].
По данным гистологического изучения трепанобиоптатов костный мозг представляется выраженно гиперклеточным за счет увеличения количества зрелых и незрелых клеток гранулоцитарного ряда и мегакариоцитов. Наибольшее количество промиелоцитов и миелоцитов располагается вблизи эндоста и периваскулярно. Мегакариоциты распределяются в срезах равномерно или образуют кластеры. По мере прогрессирования заболевания усиливается пролиферация клеток мегакариоцитарного ряда и могут выявляться многочисленные микромегакариоциты. Миелофиброз, выраженность которого усиливается по мере развития процесса, может определяться и на ранних стадиях заболевания. Увеличивается количество ретикулиновых волокон, располагающихся в виде отдельных фокусов преимущественно в периваскулярных пространствах или диффузно. Выраженность фиброза в начальной стадии развития ХМЛ коррелирует со спленомегалией, уровнем гемоглобина, процентным содержанием бластов в костном мозге и периферической крови, дополнительными кариотипическими аномалиями. Фиброз, обусловленный наличием коллагеновых волокон, встречается реже, чем вызванный ретикулиновыми нитями. Признаки остеомиелосклероза наблюдаются еще реже. Появление фиброза, переход от очагового к диффузному считается важным прогностическим фактором [9]. Выраженный фиброз обычно ассоциируется с более короткими сроками выживаемости. Но могут быть исключения. Описаны случаи ХМЛ, когда выраженный фиброз отмечался на ранних стадиях, но заболевание имело более длительное течение [10]. У ряда больных с Ph’-положительным ХМЛ могут быть выраженный фиброз за счет разрастания коллагеновых волокон и картина крови, подобная наблюдаемой при хроническом идиопатическом миелофиброзе. Поставить правильный диагноз в этих случаях помогает низкий уровень (или практически полное отсутствие) щелочной фосфатазы в нейтрофилах [1].
При ХМЛ крайне низкая активность лейкоцитарной щелочной фосфатазы (ЛЩФ) наблюдается во всех случаях — независимо от количества лейкоцитов в периферической крови и тяжести клинического течения заболевания [11]. Низкий уровень ЛЩФ обусловлен нарушением выработки фермента, а не дефектами его каталитической функции или нарушениями в образовании специфических гранул в цитоплазме лейкоцитов [12, 13]. После успешного лечения уровень ЛЩФ обычно возвращается к норме. Определение ЛЩФ, наряду с выявлением Ph’ хромосомы, используется в клинической практике для подтверждения диагноза в спорных и сомнительных случаях, когда ХМЛ приходится дифференцировать с рядом патологических процессов, сопровождающихся развитием лейкемоидных реакций миелоидного типа (инфекционные заболевания, метастазы опухолей, действие токсических веществ и др.).
К числу признаков, способствующих установлению диагноза ХМЛ (табл. 22), относится также повышенный уровень витамина В12 и витамин В12-связывающих белков в сыворотке крови больных, что, в свою очередь, обусловлено повышением содержания транскобаламинов I и II [14]. Как установлено, эти изменения, как и снижение активности ЛЩФ, увеличение количества базофилов и тромбоцитов, могут определяться за несколько лет до манифестацииХ МЛ [13].
В табл. 23 приведены клинико-лабораторные признаки, которые могут быть использованы в дифференциальной диагностике ХМЛ и лейкемоидных реакций миелоидного типа.
Иммунофенотипический анализ антигенов поверхностных мембран гранулоцитов в хронической фазе ХМЛ позволил обнаружить некоторые отличия по сравнению с клетками соответствующей степени зрелости в норме. Отмечено уменьшение плотности HLA-DR (Ia-подобного антигена) на трансформированных клетках-предшественниках гранулоцитарного ряда. На меньшем количестве клеток обнаруживался антиген CD13, увеличенным было число незрелых клеток с двойными маркерами CD15+ CD34+ [15–17], до 50% клеток реагировало с мкАТ к CD116 [17]. В целом, изучение маркеров поверхностных мембран клеток не играет существенной роли в диагностике ХМЛ в хронической фазе заболевания. При развитии же бластного криза иммунофенотипический анализ, наряду с данными цитохимических исследований, позволяет, как и при острых лейкозах, точнее идентифицировать природу бластных клеток, определяемых в костном мозге и периферической крови.
Цитогенетический анализ. Приблизительно у 5% больных с ХМЛ в лейкозных клетках при рутинном цитогенетическом исследовании не обнаруживается t (9; 22). Однако при использовании молекулярно-биологических методов и в этих случаях может быть обнаружен гибридный ген BCR/ABL. Образующийся при этом белок, обладающий повышенной активностью тирозинкиназы, может быть ответственным за усиленную пролиферацию клеток у больных с ХМЛ [2, 18–20].
Фаза акселерации и бластной трансформации
Хроническая фаза течения ХМЛ, средняя продолжительность которой составляет 3–4 года, сменяется более агрессивной и кратковременной фазой заболевания. У большинства больных развивается бластный криз (бластная трансформация), по клинико-гематологическим проявлениям близкий к острому лейкозу, развивающемуся de novo. Он характеризуется резистентностью к применяемой терапии и средней продолжительностью жизни от 3 до 6 мес [19–21]. У 10–30% больных с ХМЛ ухудшение состояния сочетается с нарастанием изменений в костном мозге и периферической крови. Но при этом количество бластов недостаточно для диагностики бластного криза. Эта стадия развития заболевания определяется как фаза акселерации, средняя продолжительность которой составляет от 12 до 18 мес [1, 3]. Сотрудниками Международного регистра по трансплантации костного мозга разработаны критерии, позволяющие четко определить переход в эти фазы заболевания [22].
Фаза бластной трансформации устанавливается при наличии не менее 30% бластов в костном мозге и периферической крови. Внезапное и быстрое увеличение содержания бластных клеток сопровождается нарастанием недостаточности костномозгового кроветворения, усилением выраженности анемии и тромбоцитопении. При бластном кризе у 70% больных патологические клетки имеют миелоидную природу, у 20–25% — лимфоидную. В 5% случаев бласты имеют дифференцировочные признаки ранних клеток-предшественников эритробластического или мегакариоцитарного ряда. Прогноз благоприятнее и чувствительность к терапии несколько выше при наличии бластов лимфоидного происхождения. Для более точного определения природы клеток при бластном кризе рекомендуется применение цитохимических и иммунологических методов [1, 2, 4, 5].
При бластной трансформации миелоидного типа клетки могут иметь цитоморфологические и цитохимические признаки миелобластов, монобластов, быть сходными с бластами при ОМЛ М4 и ОМЛ М3 [3].
При бластной трансформации лимфоидного типа клетки имеют цитологические признаки, присущие бластам при ОЛЛ L1 или L2 и очень редко ОЛЛ L3 [1, 3]. В большинстве случаев при бластном кризе лимфоидного типа клетки имеют В-клеточное происхождение и очень редко являются трансформированными ранними клетками-предшественниками Т ряда. При лимфоидном бластном кризе В-клеточного подтипа ранние клетки-предшественники имеют иммунофенотип ОЛЛ «общего типа» (ОЛЛ В II) или пре-В-клеточный (ОЛЛ В III). При бластном кризе ХМЛ лимфоидного типа чаще, чем при ОЛЛ, бластные клетки по данным иммунофенотипирования имеют бифенотипические/билинейные признаки [3]. Иногда в клинику поступают больные в стадии бластного криза, у которых по разным причинам ранее не был диагностирован ХМЛ. При этом бласты костного мозга и периферической крови могут иметь цитоморфологические и цитохимические признаки миелоидных или лимфоидных клеток-предшественников. Клинико-гематологическая картина в первом случае чаще всего напоминает ОМЛ М2, но при сохранении достаточно большого количества зрелых и незрелых гранулоцитов. При бластной трансформации лимфоидного типа не наблюдается признаков созревания TdT-положительных бластов в промиелоциты. Особые трудности возникают при дифференциальной диагностике лимфоидного бластного криза ХМЛ и Ph’-положительного ОЛЛ [1].
Почти у 30% больных с ХМЛ хроническая фаза сменяется фазой акселерации, предшествующей развитию бластного криза [1, 3]. Фаза акселерации характеризуется нарастанием миелофиброза, увеличением количества базофилов в периферической крови (>20%), снижением уровня гемоглобина (<7 г/дл). Но при этом количество бластов в костном мозге и периферической крови составляет менее 30% [21, 23]. Наблюдается также увеличение количества эозинофилов в крови и костном мозге (>10%), незрелых клеток моноцитарного ряда, эритробластов. В зрелых и незрелых миелоидных клетках обнаруживаются диспластические изменения: гиперсегментация ядер и гипогрануляция цитоплазмы нейтрофилов, различные признаки дисэритропоэза.
При исследовании трепанобиоптатов костного мозга также обнаруживаются черты дезорганизации и диспластические изменения. Увеличение количества гиподольчатых мегакариоцитов сопровождается усилением ретикулинового фиброза. Может наблюдаться коллагеновый фиброз, иногда сочетающийся с развитием остеосклероза. Увеличивается количество бластных клеток, располагающихся паратрабекулярно и периваскулярно, при одновременном уменьшении количества незрелых гранулоцитов.
Ряд больных с ХМЛ поступает в стационар в фазе акселерации, в таких случаях возникает необходимость проведения дифференциальной диагностики с различными формами МДС и острых лейкозов.
В фазе акселерации и при бластной трансформации у 80% больных с ХМЛ происходит дальнейшая эволюция кариотипа. Вторичные аномалии отмечаются чаще при миелоидном типе бластного криза, чем при лимфоидной трансформации. Основными дополнительными аномалиями, которые могут быть выявлены за несколько месяцев до развития бластного криза миелоидного типа, являются i(17q)+8, t (3; 21) (q26; q22), inv(3) (q21 q26), del(13 ) (q12 q14) [24]. Возникновение лимфоидного типа бластного криза наиболее часто сочетается с del(7) (q22) и –7.
Молекулярно-генетические аномалии, связанные с развитием бластного криза ХМЛ, касаются опухолеассоциированных супрессорных генов p53, RB1, p16 и реже онкогенов ras и myc [25, 26]. Мутации генов p16 и RB1 чаще наблюдаются в кроветворных клетках у больных с лимфоидным типом бластного криза, а гена p53 — при миелоидном типе бластной трансформации [25–27].
Атипичный миелопролиферативный синдром (Ph
–/BCR– миелолейкоз)Термины «атипичный миелопролиферативный синдром» (аМПС), «атипичный хронический лейкоз», «Ph’ отрицательный хронический миелолейкоз» употребляются для обозначения гетерогенной группы миелопролиферативных заболеваний. От ХМЛ аМПС отличается по ряду клинико-гематологических проявлений.
Основными признаками аМПС являются отсутствие Ph’-хромосомы и выявляемой реарранжировки генов BCR/ABL, гиперлейкоцитоз и нейтрофилез, наличие в периферической крови незрелых форм гранулоцитов. При аМПС, в отличие от ХМЛ, отмечаются диспластические изменения в клетках гранулоцитарного и эритробластического ряда. Количество базофилов — в пределах нормы или слегка повышено. Нечасто отмечается и эозинофилия. Количество лейкоцитов в периферической крови составляет от 20•109/л до 180•109/л. В лейкоцитарной формуле содержание промиелоцитов, миелоцитов и метамиелоцитов превышает 15%.
У больных с аМПС, а это в основном мужчины по возрасту на 15–20 лет старше, чем пациенты с ХМЛ, количество лейкоцитов в среднем ниже, но чаще наблюдается анемия и тромбоцитопения. Уровень гемоглобина колеблется в пределах от 3,4 до 14,2 г/дл. Анемия, наряду со спленомегалией, относится к числу основных жалоб при обращении за помощью к врачу-гематологу. Среднее количество тромбоцитов не превышает 80•109/л [1]. По сравнению с Ph’-положительным ХМЛ при аМПС в периферической крови увеличено относительное (но не более 10%) и абсолютное содержание моноцитов.
Диспластические изменения нейтрофилов проявляются неправильной формой ядер (псевдопельгеровские лейкоциты) и наличием гипогранулярной цитоплазмы у зрелых нейтрофилов, миелоцитов и метамиелоцитов. В большинстве случаев, как и при ХМЛ, снижен уровень активности ЛЩФ, хотя у отдельных больных может отмечаться повышенный показатель.
Костный мозг при аМПС, как правило, гиперклеточный за счет повышенного количества незрелых и зрелых клеток гранулоцитарного ряда. Но в отличие от ХМЛ лейко-эритроцитарное соотношение обычно менее 10:1. Нечасто увеличено количество незрелых базофилов и эозинофилов. Количество клеток моноцитарного ряда, напротив, увеличено. Количество мегакариоцитов у 30% больных уменьшено, в них, как и в клетках гранулоцитарного и эритробластического ряда, обнаруживаются диспластические признаки. Увеличено количество бластов, но оно не превышает 30%.
В терминальной стадии аМПС развивается бластный криз. Обнаруживаемые при этом в костном мозге и периферической крови бластные клетки чаще имеют миелоидную природу, реже относятся к клеткам-предшественникам лимфопоэза. Это служит еще одним доказательством того, что аМПС, как и ХМЛ, возникает в результате трансформации полипотентной стволовой кроветворной клетки.
Дифференциальную диагностику аМПС проводят с ХМЛ, различными формами МДС. Ряд сходных и перекрывающихся клинико-гематологических признаков имеют аМПС и такие миелопролиферативные процессы, как истинная полицитемия, эссенциальная тромбоцитемия и идиопатический миелофиброз в поздних стадиях заболевания.
Особенно сложной является дифференциация аМПС, ХМЛ и ХММЛ. ФАБ-группой предложены критерии, помогающие разграничить эти процессы [28]. Так, содержание базофилов в крови при аМПС и ХММЛ не превышает 2%, а при ХМЛ оно значительно выше. Количество моноцитов в крови больных с ХМЛ, как правило, ниже 3%, при аМПС — в пределах 3–10%, при ХММЛ — обычно выше 10%. Признаки дисплазии клеток гранулоцитарного ростка сильно выражены при аМПС, умеренно — при ХММЛ и практически не определяются при ХМЛ. Содержание незрелых гранулоцитов при аМПС колеблется в пределах 10–20%, при ХММЛ — ниже 10%, а при ХМЛ — выше 20%. Количество бластов в крови выше 2% в хронической фазе заболевания наблюдается только у больных с аМПС [1, 3]. Из числа цитогенетических аномалий при аМПС наиболее часто обнаруживается трисомия 8, моносомия 7 при отсутствии Ph’-хромосомы. Прогноз при аМПС значительно хуже, чем при ХМЛ и ХММЛ.
Хронический миеломоноцитарный лейкоз миелопролиферативного типа
Хронический миеломоноцитарный лейкоз — клональный процесс, обусловленный трансформацией полипотентной стволовой кроветворной клетки или клеток-предшественников, являющихся ее ближайшими потомками. Встречается преимущественно у лиц пожилого возраста, чаще у мужчин. Характеризуется совокупностью миелодиспластических и миелопролиферативных признаков. При преобладании первых, включая определяющуюся при исследовании периферической крови цитопению, заболевание классифицируется как одна из форм МДС — ХММЛ. Если заболевание по клинико-гематологическим проявлениям больше напоминает ХМЛ, оно обозначается как хронический миеломоноцитарный лейкоз миелопролиферативного типа (ХММЛ-МТ) [1]. Для данной формы заболевания присущи лейкоцитоз, нейтрофилез, повышенное содержание моноцитов (>3%) в периферической крови, наличие макроцитарной анемии и выраженной в различной степени спленомегалии. Абсолютное количество нейтрофилов в крови больных превышает 13•109/л, а моноцитов >1•109/л. В периферической крови обнаруживаются незрелые клетки нейтрофильного ряда. Обычно их содержание не превышает 10% от общего количества лейкоцитов. Бластные клетки встречаются редко. Изредка обнаруживают незрелые клеточные элементы моноцитарного ряда (промоноциты). Хотя это не является характерным признаком ХММЛ-МТ, но в сочетании с моноцитозом и отсутствием Ph’-хромосомы или гибридного гена BCR/ABL позволяет отличить данное заболевание от классического ХМЛ [29, 30]. Диспластические изменения в клетках гранулоцитарного ряда минимальные (псевдопельгеровские лейкоциты, нейтрофилы с гипогранулярной цитоплазмой и отрицательной реакцией при выявлении МПО) или отсутствуют. Уровень ЛЩФ у больных с ХММЛ-МТ снижен или находится в пределах нормы. Количество тромбоцитов обычно в пределах нормы или несколько уменьшено, в редких случаях отмечается тромбоцитоз.
При исследовании мазков из пунктатов отмечена гиперклеточность костного мозга с увеличением количества незрелых и зрелых клеток гранулоцитарного и в меньшей степени моноцитарного ряда. Последние составляют не менее 20% всех ядросодержащих кроветворных клеток. Клетки моноцитарного ряда представлены преимущественно моноцитами и небольшим количеством промоноцитов. Их наличие подтверждается при проведении цитохимических реакций на неспецифическую -НЭ и КНЭ [5]. При ХММЛ-МТ миелобласты и монобласты составляют не более 5% всех клеточных элементов костного мозга. Количество клеток эритробластического ряда различной степени зрелости и мегакариоцитов, как правило, в пределах нормы. Могут наблюдаться признаки дисгранулоцитопоэза и дисэритропоэза, появление гигантских многоядерных мегакариоцитов. При гистологическом изучении трепанобиоптатов костного мозга почти у 30% больных выявляется миелофиброз.
Определяемые приблизительно у трети больных цитогенетические аномалии (+8, –7 и del(20q), а также точечные мутации гена ras) не являются специфически ассоциированными с ХММЛ-МТ.
Дифференциальный диагноз ХММЛ-МТ проводят с ХМЛ, ХММЛ, лейкемоидными реакциями миелоидного типа и ОМЛ М4. Как известно, при ХММЛ-МТ в кроветворных клетках не обнаруживается Ph’ хромосома или гибридный ген BCR/ABL. Сложнее отличить ХММЛ-МТ от ХМЛ с диспластическими признаками, рассматривающегося в качестве одной из форм МДС. При этом учитывается количество лейкоцитов в периферической крови, наличие нейтрофилеза и моноцитоза, другие клинико-гематологические показатели, которые детально рассматривались выше. Проводя дифференциальную диагностику с лейкемоидными реакциями, особенно сопровождающимися моноцитозом, следует обращать внимание на их возможную связь с наличием опухолей или инфекционных заболеваний.
Течение заболевания характеризуется увеличением содержания незрелых клеток гранулоцитарного и моноцитарного ряда и прогрессирующей недостаточностью костномозгового кроветворения, возникновением осложнений, обусловленных развитием цитопении, или трансформацией в ОМЛ М4. Медиана выживаемости больных с ХММЛ-МТ ниже, чем у пациентов с ХМЛ.
Хронический нейтрофильный лейкоз
Хронический нейтрофильный лейкоз (ХНЛ) — редкое заболевание, встречающееся у лиц пожилого возраста (старше 60 лет), характеризуется наличием анемии, спленомегалии и иногда гепатомегалии. Диагноз ХНЛ устанавливают при проведении общего анализа крови, иногда случайно. Количество лейкоцитов в периферической крови колеблется в пределах 25–50•109/л и редко бывает выше 100•109/л. Преобладают сегментоядерные нейтрофильные лейкоциты (90–95%), но в отдельных случаях содержание палочкоядерных нейтрофилов составляет 20–50% [1, 3]. Крайне редко обнаруживаются незрелые гранулоциты (миелоциты, метамиелоциты) и ядросодержащие клетки эритробластического ряда, бласты отсутствуют. В некоторых нейтрофилах ядра имеют кольцевидную форму. В цитоплазме могут присутствовать токсическая зернистость и вакуоли. Миелодиспластические признаки (гипогрануляция цитоплазмы, псевдопельгеровские лейкоциты) не определяются. В лейкоцитах больных с ХНЛ, в отличие от ХМЛ, выявляется повышенный уровень щелочной фосфатазы. Морфологические признаки эритроцитов и тромбоцитов крови в пределах нормы. В сыворотке крови больных с ХНЛ, как и при ХМЛ, повышен уровень витамина В12 и витамин В12-связывающего белка.
При исследовании пунктатов и трепанатов костного мозга отмечается гиперклеточность, обусловленная выраженной гиперплазией нейтрофильных гранулоцитов. Представлены в основном зрелые и незрелые клеточные элементы этого ряда, количество миелобластов не увеличено. Признаки дисгранулоцитопоэза обычно отсутствуют. Может наблюдаться умеренное угнетение эритропоэза. Количество мегакариоцитов в пределах нормы или несколько повышено.
По цитохимическим и иммунофенотипическим признакам клетки крови и костного мозга не отличаются от соответствующих по степени зрелости гранулоцитов у здоровых людей. В кроветворных клетках при ХНЛ, в отличие от ХМЛ, не выявляются Ph’-хромосома и реарранжировка BCR/ABL. Описан ряд других цитогенетических аномалий, подтверждающих клональную природу ХНЛ, таких, как трисомия 8, трисомия 9, трисомия 21, 20q– и другие реарранжировки с вовлечением длинного плеча хромосомы 20 [31, 32].
О неопластической природе ХНЛ свидетельствуют также результаты исследований роста колоний в полутвердых средах. При этом гемопоэтические клетки-предшественники сохраняют ограниченную способность дифференцироваться исключительно в гранулоциты [33].
Важной является дифференциальная диагностика ХНЛ с нейтрофильными лейкемоидными реакциями, истинной полицитемией и хроническим идиопатическим миелофиброзом. Последние два заболевания могут также сопровождаться нейтрофилезом, а при ХНЛ в костном мозге некоторых больных могут отмечаться признаки фиброза и остеосклероза.
Прогноз при ХНЛ значительно хуже, чем у больных с ХМЛ. При прогрессировании ХНЛ в терминальный период в ряде случаев может происходить трансформация в ОМЛ [1, 3].
Гиперэозинофильный синдром/Хронический эозинофильный лейкоз
Диагноз гиперэозинофильного синдрома (ГЭС) или хронического эозинофильного лейкоза (ХЭЛ) очень трудно установить в ранней стадии заболевания. Подозрение на ГЭС/ХЭЛ возникает при наличии абсолютной эозинофилии (>1,5•109/л), удерживающейся на протяжении более 6 мес. При этом не удается установить причину эозинофилии (необходимо исключить заболевания, вызываемые паразитами, аллергию, коллагенозы, неходжкинские лимфомы, лимфогранулематоз, множественную миелому, метастазы). Развитие и созревание эозинофилов в костном мозге регулируется синергическим действием ГМ-КСФ, ИЛ-1, ИЛ-2, ИЛ-3, ИЛ-5 и другими эозинофилопоэтическими факторами, вырабатываемыми Т-лимфоцитами [2].
У больных с ГЭС/ХЭЛ могут обнаруживаться анемия, тромбоцитопения, гепатомегалия, спленомегалия, лимфаденопатия, нередко симптомы, обусловленные поражением сердца и легких. В таких случаях, если на основе цитогенетических и молекулярно-биологических исследований (включая и экспрессию гена N-ras) подтверждается клональность процесса, ставится диагноз ХЭЛ. При отсутствии этих данных диагноз ХЭЛ остается предположительным.
При исследовании периферической крови у больных общее количество лейкоцитов превышает 20•109/л. Отмечается персистирующая эозинофилия c преобладанием зрелых эозинофилов и небольшим количеством незрелых клеток. В некоторых случаях отмечаются гиперсегментация ядер, вакуолизация цитоплазмы клеток и уменьшенное содержание гранул. Аномальные гранулы или включения и палочки Ауэра не определяются. У части больных может быть умеренный нейтрофилез с небольшим количеством незрелых нейтрофилов.
Костный мозг представляется гиперклеточным за счет гиперплазии эозинофильного ростка. Представлены все стадии созревания эозинофилов, однако диспропорционального увеличения количества незрелых форм или бластов не наблюдается. В ряде случаев отмечается незначительное увеличение числа клеток нейтрофильного ряда. В эозинофилах при ХЭЛ, в отличие от нейтрофилов, не выявляется активность ХАЭ. Диспластически измененные бласты или незрелые клетки гранулоцитопоэза могут быть идентифицированы как эозинофилы на основании того, что в них определяется активность МПО, устойчивой к ингибированию цианидом. Эритропоэз и мегакариоцитопоэз не претерпевают существенных изменений.
В некоторых случаях при ХЭЛ обнаруживаются клональные хромосомные аномалии, такие как трисомия 8, i(17q), t (5; 12) (q31; q13), t (1; 5) (q23; q33) [34]. Дифференциальную диагностику проводят с Ph’ положительным ХМЛ с эозинофилией, с ХММЛ с эозинофилией, ассоциированным с t (5; 12) (q33; p13) и t (8; 13) (p11; q12), с ОЛЛ, изредка сопровождающимся эозинофилией, с ОМЛ М4Эо.
Хронический базофильный лейкоз
Базофильный лейкоз — редкое заболевание. Увеличение количества базофилов может наблюдаться в ряде случаев у больных с ХМЛ в хронической фазе и в фазе акселерации. Клетки с дифференцировочными признаками данного ряда могут наблюдаться при выделяемых в соответствии с ФАБ-классификацией подвариантах ОМЛ — М2Базо и М4Базо [3]. Базофильный лейкоз с содержанием бластов в костном мозге менее 30% имеет хроническое течение. В случаях обнаружения Ph’-хромосомы они должны рассматриваться как вариант ХМЛ [3]. При истинном хроническом базофильном лейкозе не выявляются Ph’-хромосома и реарранжировка BCR/ABL. Циркулирующие в периферической крови клетки содержат базофильные гранулы, дают отрицательную реакцию при выявлении активности МПО. В клинической картине высок удельный вес симптомов, связанных с освобождением больших количеств гистамина, наблюдается ДВС-синдром.
Хронический тучноклеточный лейкоз
Заболевание может возникнуть de novo или протекать в виде терминальной стадии системного мастоцитоза. Клиническое течение чаще хроническое или подострое, в периферической крови обнаруживаются немногочисленные тучные клетки (мастоциты). Эти атипичные клетки имеют дольчатые ядра, более высокое, чем в норме ядерно-цитоплазматическое соотношение, содержат в цитоплазме гранулы, которые метахроматически окрашиваются толуидиновым синим и дают интенсивную реакцию при цитохимическом выявлении активности ХАЭ. При исследовании мазков периферической крови могут определяться анемия, моноцитоз и эозинофилия. В эозинофилах отмечаются признаки дегрануляции и вакуолизации цитоплазмы. Костный мозг гиперклеточный за счет гиперплазии клеток гранулоцитарного ряда, эозинофилов и увеличенного количества лимфоцитов. В трепанобиоптатах костного мозга наблюдается очаговая или диффузная инфильтрация тучными клетками, гиперплазия клеток гранулоцитарного и мегакариоцитарного ряда, наличие миелофиброза и остеосклероза.
Исследования показывают, что злокачественно трансформированные тучные клетки имеют фенотип CD2+ CD4+ CD11b+ CD33+ CD8– CD19– TdT– HLA-DR–.
В клинической картине, помимо гепатомегалии, спленомегалии, лимфаденопатии, на первый план выступают симптомы, обусловленные повышенной выработкой гистамина.
Истинная полицитемия
Истинная полицитемия (ИП) (эритремия, синдром Вакеза — Ослера) — клональное миелопролиферативное заболевание, характеризующееся прежде всего избыточной продукцией клеток эритробластического ряда, а также гранулоцитов и мегакариоцитов. ИП — относительно редкое заболевание, ежегодно выявляется 5–10 новых больных на 1 млн населения. Соотношение мужчин и женщин составляет 1,2:1. Средний возраст, в котором впервые выявляется заболевание, составляет 60–70 лет. В то же время ИП иногда встречается в юношеском и даже в детском возрасте.
В своем развитии ИП проходит три последовательные стадии: фазу пролиферации, основным признаком которой является увеличение массы эритроцитов; фазу стабильного течения заболевания; фазу миелоидной метаплазии [35].
Основные критерии диагностики заболевания (табл. 24) были разработаны 25 лет тому назад группой по изучению ИП (PVSG) [36].
Диагноз ИП считается правомочным при наличии трех основных критериев категории А (А1+А2+А3) или двух первых критериев (А1+А2) и любых двух критериев категории В.
У некоторых больных для подтверждения диагноза могут использоваться и некоторые дополнительные признаки, такие, как низкий или нормальный уровень эритропоэтина в сыворотке крови (при повышенном уровне ИП исключается), рост эритроидных колоний в культуре в отсутствие эритропоэтина, клональность процесса, подтвержденная результатами цитогенетического и молекулярно-биологического исследования. Ценным является также гистологическое изучение трепанобиоптатов костного мозга, позволяющее подтвердить или исключить наличие ИП в сложных для диагностики случаях.
Симптомы, наблюдающиеся в начальной стадии заболевания, обусловлены увеличенной массой эритроцитов. Больные жалуются на слабость, головную боль, нередко в виде мучительной мигрени. У больных отмечается цианоз кожи и видимых слизистых оболочек (у 90%), боль в области сердца, в костях и нижних конечностях. У многих больных основной жалобой является кожный зуд. Нарушения со стороны ЦНС колеблются от легких функциональных (на основе стазов без тромбообразования) до тяжелых, обусловленных тромбозом крупных сосудов [37]. Тромбоз сосудов (почти у 30% больных) и кровоизлияния (у 25%) относятся к числу наиболее важных и грозных осложнений при ИП.
Одним из основных клинических симптомов у больных ИП, отнесенных к категории А, является увеличение размеров селезенки. Причиной спленомегалии, выявляющейся у 80% пациентов, является ее повышенное кровенаполнение и участие в лимфопролиферативном процессе. Почти у 70% больных одновременно обнаруживаются признаки гепатомегалии, обусловленные повышенным кровенаполнением органа, миелоидной метаплазией, разрастанием фиброзной ткани (в поздних стадиях заболевания).
Из данных лабораторных исследований основным для установления диагноза ИП является увеличение массы клеток красной крови. Присущее ИП увеличение количества эритроцитов (6–7 млн в 1 мм3) и уровня гемоглобина (18–22 г/дл) сопровождается повышением показателей гематокрита. В первой, «пролиферативной», стадии заболевания на нормобластическую эритроидную гиперплазию костного мозга указывает обнаружение в периферической крови нормохромных и нормоцитарных эритроцитов. При дефиците железа, обусловленном повышенной кровоточивостью или частыми лечебными кровопусканиями, в мазках крови могут выявляться гипохромные и микроцитарные эритроциты. В периферической крови более чем у 80% больных ИП отмечается нейтрофильный лейкоцитоз с небольшим сдвигом влево. Количество лейкоцитов обычно колеблется в пределах от 10 до 20•109/л. Умеренная базофилия наблюдается у 60–70% больных. Частой является эозинофилия.
Количество тромбоцитов повышено у 50–80% больных, у 10% из них оно выше 1000•109/л [37]. В периферической крови при ИП могут обнаруживаться крупные тромбоциты и фрагменты ядер мегакариоцитов.
В мазках костного мозга определяется гиперплазия за счет увеличения числа клеточных элементов не только нормобластического эритропоэза, но и клеток других ростков миелопоэза (гранулоцитарного, мегакариоцитарного).
Наличие гиперпластических процессов при ИП подтверждается и результатами гистологического изучения трепанобиоптатов костного мозга. У большинства больных отмечается гиперклеточность костного мозга. По данным исследователей, входящих в группу по изучению ИП, кроветворные клетки различного происхождения и разной степени зрелости занимают от 37 до 100% (в среднем 80%) всей площади срезов костного мозга [38]. Наиболее выражена гиперплазия клеток эритробластического ряда. Значительным является и увеличение количества мегакариоцитов, размер которых широко варьирует — от малых до очень крупных с дольчатыми ядрами [39]. Мегакариоциты в костном мозге распределяются в виде кластеров. В 25% случаев уже в начальной стадии ИП в костном мозге определяется увеличенное количество ретикулиновых волокон. В дальнейшем оно прогрессирует, что сочетается с увеличением клеточности костного мозга.
Длительность начальной и стабильной фаз ИП составляет от 5 до 20 лет. Примерно у 10–20% больных наблюдается переход в фазу миелоидной метаплазии. Для постполицитемической миелоидной метаплазии (ППММ) характерно уменьшение массы клеток красной крови, выявляемое при радиоизотопном исследовании, увеличение степени спленомегалии, усиление фиброза костного мозга с увеличением количества ретикулиновых и коллагеновых волокон, появление очагов экстрамедуллярного гемопоэза, развитие цитопении, пойкилоцитоз и анизоцитоз [1, 6]. Могут наблюдаться диспластические изменения в клетках эритробластического и гранулоцитарного ряда, не выявлявшиеся ранее. В ряде случаев они предшествуют развитию острого лейкоза, чаще всего миелоидного происхождения. Острый лейкоз возникает у 1–2% больных ИП, не лечившихся цитотоксическими препаратами, и у 10–15% пациентов после миелосупрессивной терапии [40]. В фазе ППММ риск развития острого лейкоза у больных значительно выше (23%), чем в начальной и стабильной фазе заболевания (7%).
Цитогенетические аномалии в момент установления диагноза выявляются почти у 40–50% больных ИП и ассоциируются с менее благоприятным прогнозом [1, 6]. Частота их увеличивается по мере прогрессирования заболевания, при эволюции в миелодиспластический процесс. Наиболее часто у больных ИП встречаются не являющиеся характерными только для этого заболевания следующие аномалии кариотипа: del(20q), +8, +9 [41]. Описания каких-либо особых изменений иммунофенотипа кроветворных клеток при ИП в доступной литературе отсутствуют.
Дифференциальную диагностику ИП проводят с другими заболеваниями, сопровождающимися вторичным (симптоматическим) эритроцитозом. Различают вторичные абсолютные эритроцитозы, при которых характерны раздражение эритробластического ростка костномозгового кроветворения и увеличение массы циркулирующих в крови эритроцитов, и относительные эритроцитозы, в основе которых лежит сгущение крови, обусловленное действием различных факторов. В онкогематологической клинике особую важность приобретает дифференциальная диагностика ИП в стабильной фазе заболевания и вторичных абсолютных эритроцитозов, встречающихся у больных с гипернефромой, опухолями почек, эндокринных органов.
ППММ и развивающийся острый лейкоз не создают диагностических проблем, так как возникают у больных ИП, длительно находившихся под наблюдением [1].
Эссенциальная тромбоцитемия
Эссенциальная тромбоцитемия (ЭТ) (синонимы: первичная тромбоцитемия, геморрагическая тромбоцитемия, идиопатическая тромбоцитемия) — миелопролиферативное заболевание, характеризующееся преимущественным поражением клеток мегакариоцитарного ряда, основным проявлением которого является выраженное увеличение количества тромбоцитов. Приводится ряд доказательств клональности процесса, обусловленного повреждением на уровне стволовой кроветворной клетки костного мозга или ее ближайших потомков [5, 42]. ЭТ относится к числу относительно редких заболеваний. Показатели ежегодной заболеваемости составляют 0,7 на 100 тыс. населения [43]. Заболевают люди в возрасте 50–70 лет, но описаны случаи развития ЭТ у лиц молодого возраста и даже у детей [44]. В клинической картине на первый план выступают осложнения, обусловленные тромбозом сосудов и кровоизлияниями. Активацией тромбоцитов и микротромбозами мелких сосудов обусловлены также часто наблюдающиеся у больных ЭТ неврологические проявления. У 50% больных определяется спленомегалия, а у 15–20% — гепатомегалия [45].
Количество тормбоцитов в периферической крови увеличено и нередко превышает 1000•109/л. Минимальным же для установления диагноза ЭТ в соответствии с критериями, предложенными группой экспертов по изучению ИП, является стойко удерживающийся в течение длительного времени уровень тромбоцитов, равный 600•109/л [46]. Уровень гемоглобина в соответствии с теми же критериями у больных ЭТ может колебаться в пределах от 10 до 18,8 г/дл (в среднем 13,8 г/дл). Среднее количество лейкоцитов в периферической крови составляет 11,5•109/л, но возможны значительные колебания — от 6 до 41•109/л. Незрелые клетки гранулоцитарного ряда в мазках периферической крови обнаруживаются крайне редко. Нет характерной для ХМЛ базофилии. Уровень щелочной фосфатазы, выявляемой при цитохимическом исследовании нейтрофилов, обычно находится в пределах нормы, в редких случаях может быть повышенным или сниженным.
Тромбоциты, выявляемые в мазках крови больных, не отличаются от наблюдающихся у здоровых людей, но могут быть полиморфными, варьируя по форме и величине. В ряде случаев могут обнаруживаться крупные, атипичные, гипогранулярные формы. Изредка в крови определяются ядросодержащие фрагменты мегакариоцитов, трудно отличимые от лимфоидных клеток. В этих случаях для идентификации природы клеточных элементов приходится прибегать к иммуноцитохимическому определению линейно-специфических антигенов — маркеров клеток мегакариоцитарного ряда, Т- и В-лимфоцитов.
У 70% больных при исследовании мазков из пунктатов и трепанобиоптатов обнаруживают гиперклеточный костный мозг. У отдельных больных количество миелокариоцитов может быть в пределах нормы или несколько увеличенным. Количество мегакариоцитов значительно повышено. Мегакариоциты в мазках равномерно распределены или образуют скопления в виде групп и кластеров. Размер мегакариоцитов с дольчатыми ядрами и обширной цитоплазмой нормальный или увеличенный. Обычно отмечается также умеренная гиперплазия клеток гранулоцитарного и эритробластического ряда. У 20–50% больных преимущественно пожилого возраста в костном мозге определяется увеличенное количество ретикулиновых волокон [46, 47].
В табл. 25 приведены критерии, позволяющие отличить ЭТ от других форм миелопролиферативных заболеваний (ИП, ХМЛ, идиопатического миелофиброза) и вторичных (реактивных) тромбоцитозов. Последние нередко отмечаются у больных с опухолями, воспалительными процессами, железодефицитной анемией, после операции спленэктомии. Следует отметить, что количество пластинок при вторичных тромбоцитозах редко превышает 1000•109/л.
Крайне сложной является дифференциальная диагностика ЭТ и ИП. Выраженный тромбоцитоз может быть также ранним проявлением ХМЛ. Лишь позднее увеличивается количество лейкоцитов и наблюдается выраженный сдвиг влево. В этих случаях помогает определение Ph’-хромосомы или обнаружение реарранжировки гена BCR/ABL.
Хронический идиопатический миелофиброз, или миелосклероз с миелоидной метаплазией, характеризуется более выраженной, чем при ЭТ спленомегалией. При гистологическом исследовании биоптатов костного мозга определяется фиброз с наличием сети коллагеновых волокон, а в крови — описанные ниже изменения.
Хронический идиопатический миелофиброз с миелоидной метаплазией
Хронический идиопатический миелофиброз с миелоидной метаплазией (ХИМММ) (синонимы; «миелосклероз с миелоидной метаплазией», «агногенная миелоидная метаплазия», «идиопатический миелофиброз», «алейкемический миелоз с остеосклерозом», «остеомиелосклероз и остеомиелофиброз») возникает в результате клональной пролиферации стволовых кроветворных клеток костного мозга [48]. Характерные признаки заболевания: панмиелоз, фиброз костного мозга и нередко остеосклероз, появление очагов экстрамедуллярного гемопоэза, спленомегалия, анемия, изменения лейкоцитарной формулы крови.
ХИМММ относится к числу редких форм миелопролиферативных заболеваний. Ежегодная заболеваемость составляет 0,5 на 100 тыс. населения [48]. Встречается преимущественно у лиц пожилого возраста (60–70 лет), но изредка и у детей [49]. Среди больных несколько преобладают лица мужского пола [50].
Основными жалобами больных при поступлении в клинику являются слабость, уменьшение массы тела, лихорадка, боль в костях, иногда геморрагические симптомы. Наиболее важными признаками ХИМММ считаются спленомегалия и гепатомегалия. В то же время у части больных первоначально отсутствуют какие-либо симптомы и заболевание выявляется случайно (на основе обнаружения увеличенной селезенки и результатов анализа периферической крови).
В момент установления диагноза может наблюдаться выраженная вариабельность гематологических параметров [51]. У большинства больных обнаруживают нормохромную анемию. У 50% больных уровень гемоглобина ниже 10 г/дл, а у 20% — 8 г/дл. Постоянными являются признаки анизоцитоза и пойкилоцитоза, наличие пойкилоцитов в форме слезинки (teardrop). У многих больных в мазках периферической крови обнаруживаются ядросодержащие клетки эритробластического ряда. Количество ретикулоцитов умеренно увеличено и может колебаться в зависимости от стадии заболевания. При наличии аутоиммунного гемолиза количество ретикулоцитов увеличивается до 5–20%.
Существенно варьирует содержание лейкоцитов в периферической крови. Изредка оно может быть низким, у 40% пациентов колеблется в пределах 10–25•109/л, у некоторых превышает 40•109/л. В крови у больных с ХИМММ встречаются гипер- или гипосегментированные лейкоциты, небольшой процент незрелых клеток гранулоцитарного ряда (миелоцитов и промиелоцитов). Уровень миелобластов, выявляемых у 40% больных, обычно не превышает 1–5%. Однако даже увеличение их количества до 10% еще не служит показателем перехода заболевания в более агрессивную фазу или трансформации в острый лейкоз [39]. Показатели лейкоцитарной щелочной фосфатазы, как правило, повышены, но у ряда больных может наблюдаться нормальный уровень или даже снижение ферментативной активности [49]. При исследовании мазков периферической крови больных ХИМММ отмечается умеренная базофилия. Количество моноцитов, как правило, не изменено. Нередко наблюдается относительная лимфоцитопения [18].
Количество тромбоцитов в периферической крови при ХИМММ может быть уменьшенным (менее 150•109/л), но может отмечаться и тромбоцитоз (более 500•109/л) [50]. В мазках периферической крови выявляются атипичные, крупные и гипогранулярные тромбоциты и аномальные мегакариоциты, в том числе микромегакариоциты, промегакариоциты и ядра мегакариоцитов. Прогрессирование заболевания сопровождается увеличением в крови количества этих клеток, нарастанием лейкоцитоза и повышением уровня незрелых клеток гранулоцитарного ряда.
Анализ миелограммы при изучении мазков костного мозга, полученных при стернальной пункции, не является показательным. Результаты разнятся в зависимости от того, попала ли игла в очаг миелоидной гиперплазии или в очаги фиброза и остеосклероза. Более информативным для установления диагноза ХИМММ, как и ряда других форм хронических миелопролиферативных заболеваний, является гистологическое изучение трепанобиоптатов костного мозга. При этом практически во всех случаях определяется гиперклеточность костного мозга, как правило, диффузная и реже очаговая. Синусы костного мозга расширены и заполнены пролиферирующими кроветворными клетками. Представлены клеточные элементы всех трех основных клеточных линий миелопоэза, хотя в отдельных срезах может быть преобладание клеток того или иного типа [1, 48]. У 90% больных отмечается выраженное количество мегакариоцитов, образующих кластеры из 3–10 клеток, характерно также наличие мегакариоцитов с конденсированными и дистрофически измененными ядрами в трепанобиоптатах с выраженным коллагеновым фиброзом.
Соотношение гемопоэтических клеток и фиброзной ткани варьирует не только в пробах, полученных из различных участков костного мозга, но и в срезах одного и того же биоптата [50]. Степень выраженности фиброза также значительно колеблется. В ранней стадии заболевания у большинства больных определяется увеличенное количество ретикулиновых волокон, располагающихся преимущественно вокруг сосудов [1, 51]. Коллагеновых волокон немного. При развитии интенсивного фиброза и возникновении очагов склероза клеточность костного мозга в отдельных участках снижается. Очажки сохранившихся клеточных элементов мегакариоцитарного, гранулоцитарного и эритробластического ряда разделены волокнами соединительной ткани. В некоторых участках костного мозга кроветворные клетки практически отсутствуют, фиброзная ткань целиком заполняет межтрабекулярные пространства.
Предпринимались попытки классификации и выделения отдельных типов ХИМММ с учетом результатов изучения трепанобиоптатов костного мозга. Так, Ward, Block [52] считают возможным выделение следующих трех основных гистологических форм: пангиперплазия, миелоидная атрофия и фиброз, миелофиброз и остеосклероз.
При пангиперплазии клетки трех основных линий миелопоэза занимают более 70% площади костного мозга. Иногда наблюдается увеличение числа ретикулиновых волокон при отсутствии коллагенового фиброза. Изредка встречаются скопления лимфоцитов. Подобная картина напоминает наблюдающуюся при ИП. При миелоидной атрофии и фиброзе небольшие очаги гемопоэза разделены ретикулиновыми и коллагеновыми волокнами. В их состав входят клетки всех основных ростков миелопоэза, но преобладают мегакариоциты. Увеличено количество плазматических клеток и клеток стромы. При третьей гистологической форме на первый план выступают признаки миелофиброза и остеосклероза. Количество гемопоэтических клеток уменьшено, и они представлены в основном мегакариоцитами.
Прогностически значимым может быть и разделение больных с ХИМММ на две группы с учетом данных гистопатологического изучения костного мозга при первоначальном установлении диагноза [51]. У больных первой группы (40%) костный мозг гиперклеточный с наличием атипичных мегакариоцитов, незрелых и зрелых гранулоцитов, клеток эритробластческого ряда, располагающихся в расширенных синусах. Ретикулиновые волокна отсутствуют или их количество несколько увеличено, располагаются они преимущественно вокруг сосудов. Коллагеновые волокна не выявляются. У больных второй группы (60%) в костном мозге отмечается умеренное или значительное уменьшение количества гемопоэтических клеток и очаги выраженного фиброза и склероза. Очажки, состоящие из незрелых клеток эритроидного и гранулоцитарного ряда, разделены участками соединительной ткани. Дистрофически измененные мегакариоциты, являющиеся преобладающими клеточными элементами, находятся в виде кластеров в расширенных синусах. У таких пациентов отмечается более низкий уровень гемоглобина и уменьшенное содержание тромбоцитов в крови, более выраженная степень спленомегалии, чем у больных первой группы.
Попутно заметим, что стромальные клетки, в первую очередь фибробласты, с которыми связывается развитие фиброза и остеосклероза, не относятся к популяции неопластических клеток. Представляется, что их пролиферация может быть связана с выработкой трансформированными мегакариоцитами повышенных количеств фактора роста фибробластов [1, 5].
Очаги экстрамедуллярного гемопоэза у больных с ХИМММ могут иметь различную локализацию, но наиболее часто возникают в селезенке, печени и лимфатических узлах [53]. В селезенке и печени клетки эритробластического, гранулоцитарного и мегакариоцитарного рядов располагаются в синусах. В лимфатических узлах, наряду с подобной же локализацией клеток трех основных ростков миелопоэза, может отмечаться выраженная перифолликулярная инфильтрация.
Цитогенетические аномалии наблюдаются почти у 60% больных с ХИМММ. Наиболее частой, хотя и неспецифической, является del 13 (q13q210). Другие возможные аномалии включают –7, +8 и +9. Кариотипические аномалии, выявляемые в момент установления диагноза, относятся к числу неблагоприятных прогностических признаков. В ряде случаев эволюция кариотипа ассоциируется с трансформацией в острый лейкоз.
Дифференциальную диагностику ХИМММ проводят со многими заболеваниями, сопровождающимися развитием фиброза и остеомиелосклероза. В первую очередь это различные формы злокачественных миелопролиферативных и лимфопролиферативных процессов. К числу первых относятся ХМЛ, ИП, ЭТ, острый миелофиброз. Для разграничения ХИМММ и ХМЛ применяют определение Ph’-хромосомы и перестройки BCR/ABL. Дифференциация ХИМММ и ИП крайне затруднена и во многих случаях должна основываться на анализе клинико-гематологических признаков.
При остром миелофиброзе отмечается агрессивное клиническое течение, отсутствие или небольшая степень спленомегалии. Помимо миелофиброза отмечаются признаки вовлечения в процесс основных ростков миелопоэза с увеличением количества незрелых и бластных клеток.
Развитием фиброза сопровождается поражение костного мозга также при неходжкинских злокачественных лимфомах. Но при этом, в отличие от ХИМММ, наблюдается мономорфная инфильтрация костного мозга клетками, соответствующими той или иной форме и цитологическому варианту исходных опухолей лимфоидной ткани.
Появление очагов специфического поражения при лимфогранулематозе также сопровождается разрастанием ретикулиновых и коллагеновых волокон. В дифференциальной диагностике решающим является обнаружение специфических для лимфогранулематоза гигантских многоядерных клеток Березовского—Штернберга.
Реактивный, или вторичный фиброз, наряду с остеосклерозом и остеолитическими изменениями, наблюдается и при метастатических поражениях костного мозга у больных раком молочной железы и других органов. В этих случаях помогает выявление раковых клеток в костном мозге на основе применения иммуноцитохимических методов и мкАТ к цитокератинам, онкофетальным антигенам, антигенам мембран эпителиальных клеток [4, 5, 54].
Развитие миелофиброза возможно также при коллагенозах, диссеминированном туберкулезе, действии ионизирующей радиации [4, 5]. Указанные заболевания и состояния также необходимо принимать во внимание при установлении диагноза ХИМММ.
Медиана выживаемости больных с ХИМММ составляет 3 года — 5 лет. Основными причинами смерти являются инфекции, кровоизлияния, тромбоэмболии. Трансформация в острый лейкоз, как правило, миелоидного происхождения отмечается у 5–20% больных [1, 48].
Хронический миелопролиферативный синдром, неклассифицируемый
Термин «хронический миелопролиферативный синдром, неклассифицируемый (ХМПС-Н)» был предложен группой по изучению ИП (PVSG) для обозначения заболевания у больных со спленомегалией, с варьирующими по степени тромбоцитозом и гиперплазией клеток мегакариоцитарного ряда, у которых не наблюдается увеличения массы эритроцитов, лейкоэритробластической реакции, значительного миелофиброза и отсутствует Ph’-хромосома [7]. Безусловно, некоторые случаи, диагностируемые как ХМПС-Н, могут быть ранними стадиями процесса, который в дальнейшем по мере получения достаточной информации удастся более точно классифицировать. Эту категорию не следует также использовать для обозначения случаев, при которых полученные при клинико-лабораторных исследованиях данные недостаточны для установления более точного диагноза.
Морфологические изменения в костном мозге и периферической крови весьма гетерогенны, но, вероятно, можно выделить следующие категории или подтипы.
Первый — случаи, когда при первоначальном исследовании костного мозга обнаруживается пангиперплазия с выраженной гиперплазией клеток мегакариоцитарного ряда, в связи с чем требуется проводить дифференциальную диагностику с ХИМММ или ИП и ЭТ. Но при этом отсутствуют обычные критерии, необходимые для установления диагноза указанных заболеваний. При исследовании периферической крови у таких больных определяют лейкоцитоз и/или выраженный тромбоцитоз. Количество эритроцитов не увеличено. Нет данных, указывающих на наличие очагов экстрамедуллярного гемопоэза.
Второй — случаи, когда начальные проявления напоминают ХИМММ (спленомегалия, лекоэритробластоз, пойкилоциты в виде слезинки, фиброз костного мозга), но при этом отмечаются выраженные диспластические изменения в клетках различных линий. Количество миелобластов существенно не повышено (<5%), в костном мозге происходит обычная дифференцировка клеток всех линий. Неясно, представляют ли такие случаи трансформацию хронической фазы ХИМММ в более агрессивное заболевание или являются «переходными» нарушениями с признаками миелопролиферативных и миелодиспластических процессов. Пока же их предлагается обозначать как ХМПС-Н.
Термин этот не может использоваться для обозначения патологических состояний с признаками, обычно наблюдающимися при хронических миелопролиферативных заболеваниях, такими, как пангиперплазия с выраженной гиперплазией мегакариоцитов, но при которых отмечаются выраженный сдвиг лейкоцитарной формулы влево с увеличением количества незрелых клеток и бластов (10–30%), миелофиброз и выраженная миелодисплазия. Последние, особенно при цитопении в периферической крови и отсутствии выраженной спленомегалии, скорее должны классифицироваться как миелодиспластические синдромы с миелофиброзом (включая «острую миелодисплазию с миелофиброзом», «острый миелосклероз» или «злокачественный миелосклероз»).
При ХМПС-Н, как и при других миелопролиферативных заболеваниях, обнаруживаются такие цитогенетические аномалии, как +8, +9, del(20q). Ph’-хромосома не определяется. Клинико-лабораторные данные у больных с ХМПС-Н вариабельны. Выделение прогностических признаков станет возможным после мониторинга течения процесса у достаточного числа больных.
Ювенильный хронический миелолейкоз (ЮХМЛ) не входит в классификацию хронических миелопролиферативных заболеваний, представленных в табл. 21. Это заболевание встречается исключительно у детей [55]. ЮХМЛ, наряду с синдромом моносомии 7, в качестве самостоятельной нозологической формы включен в классификацию миелодиспластических и миелопролиферативных заболеваний детского возраста. В ФАБ-классификации МДС [56] заболевание обозначается как ХММЛ. Этим же термином пользуются и участники Европейской рабочей группы по изучению МДС у детей [57, 58].
ЮХМЛ отличается от классического Ph’-положительного лейкоза, встречающегося в основном у взрослых, но иногда диагностируемого и у детей, по ряду морфологических, цитогенетических и клинических признаков [59, 60]. Это заболевание с агрессивным острым или подострым течением, напоминающим ОМЛ. Среди больных с ЮХМЛ 95% составляют дети (в основном мальчики) до 4 лет [55]. В момент установления диагноза возраст большинства детей не превышает 2 лет [57, 61]. Риск возникновения ЮХМЛ особенно высок у детей с семейным нейрофиброматозом [62].
Основными клиническими проявлениями заболевания являются анемия, гепатомегалия, спленомегалия, лимфаденопатия (у 20% больных), экземоподобные высыпания на коже головы, обусловленные лейкемическими инфильтратами.
У больных с ЮХМЛ умеренно снижен уровень гемоглобина. Уже в самом начале заболевания значительно уменьшается количество тромбоцитов. Общее количество лейкоцитов увеличено, но ниже, чем при ХМЛ у взрослых и колеблется в пределах 25–75•109/л [63]. При анализе лейкограммы определяют большой процент миелобластов, незрелых гранулоцитов, моноцитов, лимфоцитов и более низкий, чем при Ph’-положительном ХМЛ, уровень палочко- и сегментоядерных нейтрофильных лейкоцитов. Может определяться базофилия, не являющаяся, однако, постоянным признаком. В периферической крови могут обнаруживаться ядросодержащие клетки эритроидного ряда, количество которых увеличивается по мере прогрессирования заболевания. Встречаются отдельные плазматические клетки и иммунобласты. Костный мозг гиперклеточный, содержит увеличенное количество бластов (до 10–15%), незрелых и зрелых моноцитов. Содержание мегакариоцитов, как правило, уменьшено.
Уровень щелочной фосфатазы лейкоцитов, как и при Ph’-положительном ХМЛ, снижен. Важным лабораторным признаком является резкое увеличение (на 40–55%) уровня фетального гемоглобина у большинства больных по мере прогрессирования заболевания. При ХМЛ уровень фетального гемоглобина не превышает 10%. В качестве дополнительных признаков можно указать на наблюдающиеся при ЮХМЛ снижение гемоглобина А2 и активности карбоангидразы эритроцитов, увеличение активности глюкозо-6-фосфатдегидрогеназы и экспрессии антигена і при уменьшении экспрессии антигена І. У 50% больных повышен уровень сывороточных иммуноглобулинов [59].
У большинства больных при установлении диагноза ЮХМЛ не выявляют цитогенетических нарушений. Однако в ряде случаев описаны клональные аномалии хромосом (в основном трисомия 8), выявляемые в начальный период или в фазе прогрессирования заболевания. Частыми являются мутации онкогена ras [3].
Бластный криз развивается у небольшого числа детей. Но даже у больных, у которых не происходит бластная трансформация, прогноз неблагоприятный. Особенно тяжелое течение заболевания наблюдается у детей, заболевших в возрасте 6–12 мес. Медиана выживаемости больных с ЮХМЛ составляет 10–12 мес [3].
Дифференциальную диагностику ЮХМЛ проводят с такими заболеваниями, как редкий у детей Ph’ положительный ХМЛ, ОМЛ М4, синдром, связанный с моносомией 7, с другими миелодиспластическими/миелопролиферативными нарушениями.
1. Brunning RD, McKenna RW. Tumors of the bone marrow. Washington: Armed Forces Inst Pathol 1993; 406 p.
2. Schumacher HR, Coteligham JD. Chronic leukemia. Approach to diagnosis. New York: Igaku-Shoin Med Publ 1993; 356 p.
3. Bain BJ. Leukemia Diagnosis. 2nd ed. Oxford: Blackwell Science 1999. 200 p.
4. Hoffbrand AV, Pettit JE. Color atlas of hematology. 2nd ed. London etc: Mosby-Wolfe 1998. 360 p.
5. Глузман ДФ, Абраменко ИВ, Скляренко ЛМ, Надгорная ВА. Лабораторная диагностика онкогематологических заболеваний. Киев: Морион 1998. 336 с.
6. Neoplastic Hematopathology. Ed by DM Knowles, Baltimore et al: Williams a Wilkins 1992; 1624 p.
7. Imbert M, Pierre R, Vardiman J. Chronic Myeloproliferative Disorders. Writting Committee. Chicago 1998; 1–12.
8. Morrison VA. Chronic leukemias. Ca-A Cancer J Clin 1994; 44: 353.
9. Dekmezian R, Kantarjian HM, Keating MY et al. The relevance of reticulin stain-measured fibrosis at diagnosis in chronic myelogenous leukemia. Cancer 1987; 59: 1739.
10. Clough V, Geary CG, Hashmi K et al. Myelofibrosis in chronic granulocytic leukaemia. Br J Haematol 1979; 42: 515.
11. Глузман ДФ. Диагностическая цитохимия гемобластозов. Киев: Наук думка 1978; 216 с.
12. Wachstein M. Histochemistry of enzymes in tumors. Handbuch der Histochemie, Bd 2. 1962: 73.
13. Kamada N, Uchino H. Chronologic sequence in appearance of clinical and laboratory findings characteristic of chronic myelocytic leukemia. Blood 1978; 51: 843.
14. Zittoun J, Zittoun R, Marquet J, Sultan C. The three transcobalamins in myeloproliferative disorders and acute leukaemia. Br J Haematol 1975; 31: 287.
15. Todd MB, Waldron JA, Jennings TA et al. Loss of myeloid differentiation antigens precledes blastic transformation in chronic myelogenous leukemia. Blood 1987; 70: 122.
16. Schmetzer HM, Gerhartz HH. Immunological classification of chronic phase imminent blastic transformation and acute lymphoblastic leukemia. Exp Hematol 1997; 6: 502.
17. Логинский ВЕ, Выговская ЯИ, Потемкина ГИ и др. Иммунофенотипирование мононуклеарных клеток при хронических миелопролиферативных заболеваниях. Эксперим онкология 1991; 13: 37.
18. Wardiman JW. Chronic myelogenous leukemia and myeloproliferative disorders. In: Neoplastic Hematopathology 1992: 1405.
19. Lichtman MA. Chronic myelogenous leukemia and related disorders. In: Williams Hematology. 5th ed. 1995: 298.
20. Deininger MW, Goldman JM. Chronic myeloid leukemia. Curr Opin Hematol 1998; 5: 302.
21. Katarjian HM, Deisseroth A, Kurzrock R et al. Chronic myelogenous leukemia: a coincise update. Blood 1993; 82: 691.
22. Savage DG, Szydlo RM, Chase A et al. Bone marrow transplantation for chronic myeloid leukemia: the effects of differing criteria for defining chronic phase on probabilities of survival and relapse. Br J Haematol 1997; 99: 30.
23. Muehleck SD, McKenna RW, Arthur DC et al. Transformation of chronic myelogenous leukemia: clinical, morphologic and cytogenetic features. Am J Clin Pathol 1984; 82: 1.
24. Champlin RE, Golde DW. Chronic myelogenous leukemia: recent advances. Blood 1985; 65: 1039.
25. Ahuja HG, Jat PS, Foti A et al. Abnormalities of the retinoblastoma gene in the pathogenesis of acute leukemia. Blood 1991; 78: 3259.
26. Ishikura H , Yufu Y, Yamashita S et al. Biphenotypic blast crisis of chronic myelogenous leukemia: abnormalities of p53 and retinoblastoma genes. Leuk Lymphoma 1997; 25: 573.
27. Sill H, Goldman JM, Gross NCP. Homozygous deletion of the p16 tumor-suppressor gene are associated with lymphoid transformation of chronic myeloid leukemia. Blood 1995; 85: 2013.
28. Bennet JM, Catovsky D, Daniel MT et al. The chronic myeloid leukemias: guidelines for distinguishing chronic granulocytic, atypical myeloid, and chronic myelomonocytic leukemia. Proposals by the French-American-British Cooperative group. Br J Haematol 1994; 87: 746.
29. Pugh WC, Pearson M, Vardiman JW, Rowley JD. Philadelphia chromosome-negative chronic myelogenous leukaemia: a morphologic reassessment. Br J Haematol 1985; 60: 457.
30. Kantarjian HM, Kurzrock R, Talpaz M. Philadelphia chromosome-negative chronic myelogenous leukemia and chronic myelomonocytic leukemia. Hematol Oncol Clin North Am 1990; 4: 389.
31. Hasle H, Olesen G, Kerndrup G et al. Chronic neutrophilic leukemia in adolescence and young adulthood. Br J Haematol 1996; 94: 628.
32. Matano S, Nakamura S, Kobayashi K et al. Deletion of the long arm of chromosome 20 in a patient with chronic neutrophilic leukemia: cytogenetic findings in chronic neutrophilic leukemia. Am J Hematol 1997; 54: 72.
33. Yanagisava K, Ohminami H, Sato M et al. Neoplastic involvement of granulocytic-monocytic, or erythrocytic lineage in a patient with chronic neutrophilic leukemia. Am J Hematol 1998; 57: 221.
34. Bain BJ. Eosinophilic leukaemias and the idiopathic hypereosinophilic syndrome. Br J Haematol 1996; 95: 2.
35. Beutler E. Polycythemia vera. In: Williams Hematology. 5th ed. 1995; 324.
36. Berlin NJ. Diagnosis and classification of the polycythemias. Semin Hematol 1975; 12: 339.
37. Berk PD, Goldberg JD, Donovan PB et al. Therapeutic recommendation in polycythemia vera based on Polycythemia Vera Study Group protocols. Semin Hematol 1986; 23: 132.
38. Ellis JT, Peterson P, Geller SA, Rappaport H. Studies of the bone marrow in polycythemia vera and the evolution of myelofibrosis and secondary hematologic malignancies. Semin Hematol 1986; 23: 144.
39. Vykoupil KF, Thiele J, Stangel W et al. Histopathology, ultrastructure and cytogenetics of the bone marrow in comparison with secondary polycythemia. Virchovs Arch A 1980; 389: 307.
40. Landaw SA. Acute leukemia in polycythemia vera. Semin Hematol 1986; 23: 156.
41. Diez-Martin JL, Graham DL, Petitt RM et al. Chromosome studies in 104 patients with polycythemia vera. Mayo Clin Proc 1991; 66: 287.
42. Fialkow PY, Faquet GB, Jacobson RJ et al. Evidence that essential thrombocythemia is a clonal disorder with origin in a multipotent stem cell. Blood 1981; 58: 916.
43. McIntyre KJ, Hoagland HC, Silverstein MN, Petitt RM. Essential thrombocythemia in young adults. Mayo Clin Proc 1991; 66: 149.
44. Mitus AJ, Schsfer AJ. Thrombocytosis and thrombocythemia. Hematol Oncol Clin North Am 1990; 4: 157.
45. Schafer AJ. Essential (primary) trombocythemia. In: Williams Hematology 5th ed. 1995; 340.
46. Murphy S, Iland H, Rosental D, Laszlo J. Essential thrombocythemia: an interim report from the Polycythemia Vera Study Group. Semin Hematol 1986; 23: 177.
47. Wintrobes Clinical Haematology, 10th ed. Baltimore: Williams a Wilkins 1999; 2764 p.
48. Lichtman MA. Idiopathic myelofibrosis (agnogenic myeloid metaplasia). In: Williams Hematology 5th ed. 1995; 311.
49. Weinstein IM. Idiopathic myelofibrosis: Historical review, diagnosis and management. Blood Rev 1991; 5: 98.
50. Visani G, Finelli C, Castelli U et al. Myelofibrosis with myeloid metaplasia: clinical and haematological parameters predicting survival in a series of 133 patients. Br J Haematol 1990; 75: 4.
51. Thielle J, Zankovich R, Steinberg T et al. Agnogenic myeloid metaplasia (AMM) — correlation of bone marrow lesions with laboratory data: a longitudinal clinicopathological study of 114 patients. Hematol Oncol 1989; 7: 327.
52. Ward HP, Block MH. The natural history of agnogenic myeloid metaplasia (AMM) and a critical evaluation of its relationship with myeloproliferative syndrome. Medicine 1971; 50: 357.
53. Beckman EN, Oehrle JS. Fibrous hematopoietic tumors arising in agnogenic myeloid metaplasia. Hum Pathol 1982; 13: 804.
54. Cripe LD, Hromas RW. Malignant disorders of megakaryocytes. Semin Hematol 1998; 35: 200.
55. Passmore SJ, Hann JM, Stiller CA et al. Pediatric myelodysplasia: a study of 68 children with a new prognostic scoring system. Blood 1995; 85: 1742.
56. Bennet JM, Catovsky D, Daniel MT et al. Proposed revised criteria for the classification of acute myeloid leukemia. Ann Intern Med 1985; 103: 626.
57. Neimeyer CM, Arico M, Basso G et al. Chronic myelomonocytic leukemia in childhood: a retrospective analysis of 110 cases. Blood 1997; 89: 3534.
58. Neimeyer CM, Fenu S, Hasle H et al. Differentiating juvenile myelomonocytic leukemia from infections disease. Blood 1998; 91: 365.
59. Smith KL, Johnson W. Classification of chronic myelocytic leukemia in children. Cancer 1974; 34: 670.
60. Freedman MH, Estrov Z, Chan HS. Juvenile chronic myelogenous leukemia. Am J Pediatr Hematol Oncol 1988; 10: 261.
61. Arico M, Biondi A, Pui CH. Juvenile myelomonocytic leukemia. Blood 1997; 90: 479.
62. Clark RD, Hutter JJ. Familial neurofibromatosis and juvenile chronic myelogenous leukemia. Hum Genet 1982; 60: 230.
63. Hardisty RM, Speed DE, Till M. Granulocytic leukemia in childhood. Br J Haematol 1964; 10: 551.
Разница между острым и хроническим лейкозом
Лейкоз — это рак, поражающий кровь, лимфатическую систему и костный мозг, мягкую внутреннюю часть костей, где образуются новые стволовые клетки крови. Обычно стволовые клетки постепенно превращаются в здоровые эритроциты, лейкоциты и тромбоциты, которые циркулируют по всему телу и выполняют многие жизненно важные функции, такие как транспортировка кислорода и питательных веществ.
Лейкоз развивается при повреждении ДНК развивающихся клеток крови.В результате клетки крови бесконтрольно растут и делятся, вытесняя здоровые клетки крови из костного мозга и потенциально распространяясь на другие части тела.
Типы лейкемии
Лейкоз — это сложная форма рака, которая имеет несколько типов, которые определяются на основе пораженных клеток крови (миелоидных или лимфоидных) и скорости прогрессирования рака (острого или хронического).
Острый лейкоз
Острый лейкоз препятствует созреванию стволовых клеток крови.Состояние очень быстро прогрессирует, образуя большое количество аномальных лейкоцитов, которые не функционируют должным образом.
Симптомы острого лейкоза, которые, как правило, появляются раньше и являются более серьезными, чем симптомы хронического лейкоза, могут включать:
- Необъяснимая усталость
- Непреднамеренная потеря веса
- Лихорадка без очевидной причины
- Одышка
- Кожа бледная (от анемии)
- Маленькие красные пятна под кожей
- Частые инфекции, не поддающиеся лечению
- Легкие синяки и кровотечения
- Медленно заживающие раны
- Боль в костях и суставах
Хронический лейкоз
Хронический лейкоз подавляет развитие стволовых клеток крови, в конечном итоге заставляя их функционировать менее эффективно, чем здоровые зрелые клетки крови.По сравнению с острым лейкозом хронический лейкоз имеет тенденцию быть менее тяжелым и прогрессировать медленнее. По мере распространения рака могут появиться такие симптомы, как:
- Шишки на шее, подмышках или паху (вызванные увеличением лимфатических узлов)
- Боль или ощущение переполнения в животе (из-за увеличения селезенки или печени)
- Лихорадка, озноб и ночная потливость
- Слабость и утомляемость
Лечение лейкемии зависит от ее типа.Поэтому ранний и точный диагноз важен для достижения наилучшего результата и качества жизни.
Проверено с медицинской точки зрения Четаси Талати, доктором медицины, злокачественной гематологии.
Если вы хотите поговорить со специалистом по лейкемии из программы по злокачественной гематологии онкологического центра Моффитта, позвоните по телефону 1-888-663-3488 или заполните нашу новую форму регистрации пациента онлайн.
типов лейкемии: общие, редкие и другие разновидности
Лейкоз классифицируется по типу пораженных лейкоцитов и по скорости прогрессирования заболевания.Лимфоцитарный лейкоз (также известный как лимфоидный или лимфобластный лейкоз) развивается в белых кровяных тельцах, называемых лимфоцитами, в костном мозге. Миелоидный (также известный как миелогенный) лейкоз может также начаться в белых кровяных тельцах, помимо лимфоцитов, а также в красных кровяных тельцах и тромбоцитах.
С точки зрения скорости развития или ухудшения лейкемии лейкоз подразделяется на острый (быстрорастущий) или хронический (медленнорастущий). Острый лейкоз быстро прогрессирует и приводит к накоплению незрелых, лишенных функций клеток крови в костном мозге.При этом типе лейкемии клетки воспроизводятся и накапливаются в костном мозге, снижая способность костного мозга производить достаточно здоровых клеток крови. Хронический лейкоз прогрессирует медленнее и приводит к накоплению относительно зрелых, но все еще аномальных лейкоцитов.
Типы лейкемии включают:
Острый лимфоцитарный лейкоз (ОЛЛ) быстро прогрессирует, заменяя здоровые клетки, производящие функциональные лимфоциты, лейкозными клетками, которые не могут созреть должным образом.Клетки лейкемии переносятся с кровотоком в другие органы и ткани, включая мозг, печень, лимфатические узлы и яички, где они продолжают расти и делиться. Рост, деление и распространение этих лейкозных клеток может привести к ряду возможных симптомов.
Узнать больше про ВСЕ
Острый миелоидный лейкоз (AML) , также известный как острый миелогенный лейкоз, острый миелобластный лейкоз, острый гранулоцитарный лейкоз или острый нелимфоцитарный лейкоз, представляет собой быстрорастущую форму рака крови и костного мозга.
Подробнее о AML
Хронический лимфолейкоз (ХЛЛ) — это обычно медленно растущий рак, который начинается в лимфоцитах костного мозга и распространяется в кровь. Он также может распространяться на лимфатические узлы и органы, такие как печень и селезенка. ХЛЛ развивается, когда растет слишком много аномальных лимфоцитов, вытесняя нормальные клетки крови и затрудняя организму борьбу с инфекцией.
Подробнее о CLL
Хронический миелоидный лейкоз (ХМЛ) , также известный как хронический миелолейкоз, начинается в кроветворных клетках костного мозга и затем со временем распространяется в кровь.Со временем болезнь распространяется на другие части тела.
Подробнее о CML
Волосатоклеточный лейкоз (HCL) — редкий подтип хронического лимфоцитарного лейкоза (CLL), который прогрессирует медленно. HCL возникает, когда костный мозг производит слишком много B-клеток (лимфоцитов), типа белых кровяных телец, которые борются с инфекцией. По мере увеличения количества лейкозных клеток вырабатывается меньше здоровых лейкоцитов, красных кровяных телец и тромбоцитов.
Подробнее о HCL
Миелодиспластические синдромы (МДС) — это группа тесно связанных заболеваний, при которых костный мозг производит слишком мало функционирующих эритроцитов (которые переносят кислород), лейкоцитов (которые борются с инфекцией) или тромбоцитов (которые предотвращают или останавливают кровотечение ) или любую их комбинацию.Различные типы миелодиспластических синдромов диагностируются на основании определенных изменений в клетках крови и костном мозге. Клетки в крови и костном мозге (также называемые миело) обычно выглядят ненормально (или диспластичными), отсюда и название миелодиспластических синдромов.
По данным Американского онкологического общества, ежегодно около 13 000 человек заболевают МДС. В прошлом МДС обычно называли прелейкемическим состоянием (и его до сих пор иногда называют прелейкемией), потому что у некоторых людей с МДС развивается острый лейкоз как осложнение заболевания.Однако у большинства пациентов с МДС никогда не разовьется острый лейкоз.
По соглашению, МДС классифицируется как острый миелоидный лейкоз (ОМЛ) с миелодиспластическими особенностями, когда бласты крови или костного мозга достигают или превышают 20 процентов.
Узнайте больше о признаках, симптомах и методах лечения рака крови.
Следующая тема: Что такое острый лимфолейкоз?
Что такое хронический миелоидный лейкоз?
Рак начинается, когда клетки организма начинают бесконтрольно расти.Клетки практически в любой части тела могут стать раком и распространиться на другие части тела. Чтобы узнать больше о том, как рак начинается и распространяется, см. Что такое рак?
Хронический миелолейкоз (ХМЛ) также известен как хронический миелолейкоз . Это тип рака, который начинается в определенных кроветворных клетках костного мозга.
При ХМЛ генетические изменения происходят в ранней (незрелой) версии миелоидных клеток — клеток, вырабатывающих эритроциты, тромбоциты и большинство типов белых кровяных телец (кроме лимфоцитов).Это изменение формирует аномальный ген под названием BCR-ABL , который превращает клетку в клетку CML. Клетки лейкемии растут и делятся, накапливаясь в костном мозге и попадая в кровь. Со временем клетки могут оседать и в других частях тела, включая селезенку. ХМЛ — это довольно медленно растущий лейкоз, но он может превратиться в быстрорастущий острый лейкоз, который трудно лечить.
ХМЛ чаще встречается у взрослых, но очень редко встречается и у детей. В целом лечение у них такое же, как и у взрослых.
Что такое лейкемия?
Лейкоз — это рак, который начинается в кроветворных клетках костного мозга. Когда одна из этих клеток изменяется и становится лейкозной клеткой, она перестает созревать должным образом. Часто он делится, чтобы новые клетки производились быстрее, чем обычно. Клетки лейкемии также не умирают, когда должны. Они накапливаются в костном мозге и вытесняют нормальные клетки. В какой-то момент лейкозные клетки покидают костный мозг и попадают в кровоток, часто вызывая увеличение количества лейкоцитов (лейкоцитов) в крови.Попадая в кровь, лейкозные клетки могут распространяться на другие органы, где они могут препятствовать нормальной работе других клеток в организме.
Лейкемия отличается от других видов рака, которые начинаются в таких органах, как легкие, толстая кишка или грудь, а затем распространяются на костный мозг. Рак, который начинается в другой части тела, а затем распространяется на костный мозг, не является лейкемией.
Не все лейкозы одинаковы. Знание конкретного типа лейкемии помогает врачам лучше предсказать прогноз (перспективы) каждого пациента и спланировать лучшее лечение.
Что такое хронический лейкоз?
Лейкоз — это острый или хронический в зависимости от того, являются ли большинство аномальных клеток незрелыми (и больше похожи на стволовые клетки) или зрелыми (и больше похожи на нормальные лейкоциты).
При хроническом лейкозе клетки созревают частично, но не полностью. Эти клетки могут выглядеть довольно нормально, но это не так. Как правило, они не борются с инфекцией так же хорошо, как нормальные лейкоциты. Клетки лейкемии также живут дольше, чем нормальные клетки, накапливаются и вытесняют нормальные клетки в костном мозге.Хроническим лейкозам может пройти много времени, прежде чем они вызовут проблемы, и большинство людей могут жить долгие годы. Но хронические лейкозы, как правило, труднее вылечить, чем острые лейкемии.
Что такое миелоидный лейкоз?
Является ли лейкоз миелоидным или лимфоцитарным , зависит от того, в каких клетках костного мозга начинается рак.
- Миелоидные лейкозы (также известные как миелоцитарные , миелогенные или нелимфоцитарные лейкозы) начинаются в ранних миелоидных клетках — клетках, которые становятся лейкоцитами (кроме лимфоцитов), эритроцитами или тромбоцитами. -образующие клетки (мегакариоциты).
- Лимфоцитарный (также известный как лимфоидный или лимфобластный лейкоз) начинается в клетках, которые становятся лимфоцитами.
Какие еще типы лейкемии?
Существует 4 основных типа лейкемии, в зависимости от того, являются ли они острыми или хроническими, а также миелоидные или лимфоцитарные:
При остром лейкозе клетки костного мозга не могут созревать должным образом. Эти незрелые клетки продолжают воспроизводиться и накапливаться.Без лечения большинство людей с острым лейкозом проживут всего несколько месяцев. Некоторые виды острого лейкоза хорошо поддаются лечению, и многие пациенты излечиваются. Другие виды острого лейкоза имеют менее благоприятный прогноз.
Лимфоцитарный лейкоз начинается в клетках, которые становятся лимфоцитами. Лимфомы также являются раком, который начинается в этих клетках. Основное различие между лимфолейкозом и лимфомой заключается в том, что при лейкозе раковые клетки находятся в основном в костном мозге и крови, тогда как при лимфомах они, как правило, находятся в лимфатических узлах и других тканях.
Хронический миеломоноцитарный лейкоз (ХММЛ) — еще один хронический лейкоз, который начинается в миелоидных клетках. Для получения дополнительной информации см. Хронический миеломоноцитарный лейкоз.
Какие типы лейкемии?
Врачи классифицируют лейкоз в зависимости от типа лейкоцитов — лимфоцитов или миелоидных клеток — и от того, развивается ли болезнь очень быстро ( острое заболевание ) или медленно ( хроническое заболевание ).
Лимфоцитарные лейкозы развиваются из клеток, которые дают начало Т-лимфоцитам (Т-клеткам), В-лимфоцитам (В-клетки) или естественным киллерам (NK). Каждый из этих типов клеток играет особую роль в иммунной системе; одни вырабатывают антитела, тогда как другие напрямую борются с другими иммунными клетками или направляют их на борьбу с инфекциями.
Миелоидные лейкозы развиваются из клеток, дающих начало лейкоцитам, называемым гранулоцитами и моноцитами. Гранулоциты получили свое название от гранул, содержащих ферменты, которые они несут внутри себя.Они высвобождают эти ферменты при встрече с бактериями или грибками. Моноциты в конечном итоге становятся макрофагами, которые поглощают и уничтожают бактерии и грибки.
При остром лейкозе, который развивается быстро, злокачественные клетки (называемые бластами ) незрелые и неспособны выполнять свои функции иммунной системы. Хронические лейкозы развиваются в более зрелых клетках, которые могут выполнять некоторые из своих функций, но не очень хорошо. Эти аномальные клетки обычно размножаются медленнее, чем при остром лейкозе.
Из четырех распространенных типов лейкемии у взрослых наиболее часто встречаются острый миелоидный лейкоз (ОМЛ) и хронический лимфоцитарный лейкоз (ХЛЛ). Другие родственные виды рака крови включают миелопролиферативные новообразования и системный мастоцитоз.
- О волосатоклеточном лейкозе (HCL)
Волосатоклеточный лейкоз (HCL) — это редкий вид рака у взрослых, который обычно обнаруживается во время тестирования на низкое содержание крови.Узнайте больше о том, что врачи MSK ищут при диагностике HCL.
- Острый лимфоцитарный лейкоз (ОЛЛ)
Острый лимфоцитарный лейкоз (ОЛЛ) — редкая форма рака у взрослых, но наиболее распространенная форма лейкемии у детей. Узнайте больше о том, что врачи MSK ищут при постановке диагноза ВСЕ.
- Острый миелоидный лейкоз (ОМЛ)
Острый миелоидный лейкоз (ОМЛ) — один из наиболее распространенных лейкозов у взрослых.Узнайте больше о AML и способах его диагностики.
- Острый промиелоцитарный лейкоз (APL)
Острый промиелоцитарный лейкоз (APL) — это агрессивный тип острого миелоидного лейкоза. Узнайте больше об APL и способах его диагностики.
- Хронический лимфоцитарный лейкоз (ХЛЛ)
Хронический лимфолейкоз (ХЛЛ) является наиболее распространенным хроническим лейкозом у взрослых.Методы лечения ХЛЛ быстро улучшаются и меняются. Узнайте о методах лечения ХЛЛ.
- Хронический миелоидный лейкоз (ХМЛ)
Узнайте больше о том, какую роль хромосомы играют в развитии хронического миелоидного лейкоза (ХМЛ).
- Миелопролиферативные новообразования (MPN)
Узнайте, что делает костный мозг и как это может привести к развитию заболеваний, известных как миелопролиферативные новообразования.
- Системный мастоцитоз
Мастоцитоз возникает, когда в организме накапливается слишком много тучных клеток. Прочтите о том, как диагностируется это расстройство и его связь с аллергией.
Чем ХЛЛ отличается от других типов лейкемии
Хронический лимфолейкоз (ХЛЛ) начинается в костном мозге, мягкой внутренней части ваших костей. Есть некоторые ключевые отличия, которые отличают ХЛЛ от других типов лейкемии, таких как ОЛЛ (острый лимфолейкоз), ОМЛ (острый миелоидный лейкоз) и ХМЛ (хронический миелоидный лейкоз).
Кто заболеет
ХЛЛ — наиболее распространенная лейкемия у взрослых. Большинству людей диагноз ставят в возрасте от 60 до 70 лет. С возрастом ваш риск увеличивается. Другие типы, такие как ОЛЛ и ОМЛ, в основном возникают в детстве. Дети обычно не болеют медленнорастущими формами лейкемии.
Большинство людей живут с ХЛЛ намного дольше, чем с другими видами рака или другими видами лейкемии.
Где это начинается
ХЛЛ и ВСЕ начинаются в костном мозге, где обычно образуются клетки крови. В частности, он начинается в клетке костного мозга, называемой лимфоцитами.(Это то, что обозначает первая буква «L» в словах CLL и ALL.)
Лимфоциты становятся разновидностью лейкоцитов, которые помогают вашему организму бороться с инфекциями. Но при ХЛЛ лимфоциты бесконтрольно разрастаются. Анализы крови часто показывают высокий уровень лейкоцитов или лимфоцитов. Вам нужно будет пройти еще несколько анализов, чтобы узнать, лейкемия ли это.
Два других типа лейкемии — острый миелоидный лейкоз (ОМЛ) и хронический миелоидный лейкоз (ХМЛ) — начинаются в миелоидных клетках костного мозга. Миелоидные клетки развиваются в белые кровяные тельца, отличные от лимфоцитов, красные кровяные тельца (которые переносят кислород) и тромбоциты (которые образуют сгустки, помогающие остановить кровотечение).
Как быстро растут раковые клетки
Буква «C» в CLL означает хронический. Это означает, что рак обычно растет и распространяется медленно, хотя ХЛЛ также может расти быстрее.
Напротив, типы лейкозов, в названии которых есть слово «острый» (острый лимфоцитарный лейкоз и острый миелоидный лейкоз), растут намного быстрее, чем большинство хронических лейкозов. Люди с этим типом обычно очень больны и нуждаются в немедленном лечении.
Если, как и у большинства людей с ХЛЛ, у вас медленнорастущий тип, вы можете не замечать никаких симптомов в течение многих лет.Обычный анализ крови может быть первым признаком того, что что-то не так. Вам может не понадобиться лечение сразу. (Ваш врач, конечно, будет внимательно следить за вашим здоровьем.) Нужно ли вам начинать лечение, зависит от ваших симптомов, результатов анализов и стадии (от 0 до IV) вашего заболевания.
Врачи могут проверять общие и необычные белки на поверхности клеток ХЛЛ. Результаты помогают диагностировать и лечить его, а также проверять прогноз (как это состояние может повлиять на вас).
В чем разница между острым и хроническим лейкозом? Онкологический центр
Стволовые клетки происходят из нашего костного мозга и в процессе роста становятся эритроцитами, лейкоцитами и тромбоцитами.Эти клетки составляют нашу кровь и играют жизненно важную роль в нашем здоровье, доставляя кислород к нашим органам, помогая бороться с инфекциями и останавливая кровотечение в случае порезов.
Если эти клетки останавливаются в своем развитии, это может привести к лейкемии. В зависимости от типа поврежденных стволовых клеток и того, когда происходит нарушение их роста, лейкоз можно разделить на две группы: острые и хронические.
Наш Элиас Джаббур, доктор медицины, рассказывает о различиях между острыми и хроническими лейкозами, в том числе о том, как они делятся на подтипы и как это помогает в выборе лечения.
В чем разница между острым и хроническим лейкозом?
Как и все живые существа, клетки имеют жизненный цикл: они рождаются, созревают и затем умирают. У лейкемии этот жизненный цикл заблокирован. Детские клетки не созревают или взрослые клетки никогда не умирают. Лейкемии классифицируются в зависимости от того, когда клетки отклоняются от своего жизненного цикла.
- Хронический лейкоз возникает в результате сбоя в жизненном цикле после созревания клеток.Взрослые клетки часто в некоторой степени все еще могут функционировать, но, в конечном счете, они несовместимы с жизнью.
- Острый лейкоз возникает при остановке развития на ранних этапах жизненного цикла клетки. Эти детские клетки, находящиеся в тупике, не имеют никакой функции.
Как лимфоидные или миелоидные клетки подходят для диагностики лейкемии?
Помимо хронического и острого, диагноз лейкемии может быть дополнительно определен путем определения того, какой из двух типов стволовых клеток поражен: лимфоидные клетки или миелоидные клетки.
Лимфоидные клетки производят лимфоциты — белые кровяные тельца, составляющие большую часть иммунной системы. Миелоидные клетки производят эритроциты, тромбоциты и другие типы белых кровяных телец, не являющиеся лимфоцитами.
Хотя существуют более редкие формы болезни, большинство лейкозов делятся на четыре основных подтипа:
- острый миелоидный лейкоз
- Острый лимфобластный лейкоз
- хронический миелолейкоз
- хронический лимфолейкоз
Независимо от того, когда в жизненном цикле происходит сбой и какие клетки поражены, раковые клетки вытесняют здоровые клетки и мешают им выполнять свою важную работу.
Различаются ли симптомы при хроническом и остром лейкозе?
Да, хронические лейкозы развиваются медленно, поэтому у большинства пациентов не так много симптомов.
Однако пациенты с острыми лейкозами обычно легко кровоточат, например, при чистке зубов. У них могут быть частые кровотечения из носа или обильные менструации. Также они часто испытывают жар и утомляемость.
Как лечат острый лейкоз?
Цель лечения — восстановить нормальное функционирование клеток.При остром лейкозе первый шаг называется индукцией. С помощью химиотерапии мы лечим раковые клетки крови, а также костный мозг. Чтобы убедиться, что все раковые клетки исчезли, существует второй этап лечения, называемый консолидацией. На этом этапе некоторые пациенты получают больше химиотерапии, а другие получают больше химиотерапии плюс трансплантацию стволовых клеток. Подход консолидации зависит от того, насколько агрессивен лейкоз, а также от уникальных генетических характеристик диагноза, который определяется с помощью серии тестов при первоначальном диагнозе.
Третий этап — это обслуживание, которое помогает снизить риск рецидива рака. В течение двух-трех лет пациенты принимают химиотерапию в основном в виде таблеток и попадают в больницу только раз в месяц. Мы использовали этот подход в течение некоторого времени для лечения острого лимфобластного лейкоза, но новое исследование показывает, что он помогает продлить жизнь и пациентам с острым миелоидным лейкозом.
Наконец, поскольку некоторые химиотерапевтические препараты не проходят через так называемый гематоэнцефалический барьер, мы использовали лучевую терапию, чтобы предотвратить рецидив.Но это может вызвать долгосрочные побочные эффекты, которые могут иметь большое значение для наших педиатрических пациентов с лейкемией. Теперь мы используем интратекальную химиотерапию, которая вводится в позвоночник и распространяется до мозга.
Как лечат хронические лейкозы?
Любой диагноз рака — проблема, но хорошая новость заключается в том, что с хроническим лейкозом вы можете жить нормальной жизнью. Фактически, 40% пациентов с хроническим лимфолейкозом можно просто наблюдать и не нуждаться в лечении.
Когда приходит время лечить рак, мы используем таргетную терапию, поскольку пациенты с хроническими лейкозами, как правило, старше и имеют больше данных в истории болезни.
Химиотерапия убивает все в надежде, что раковые клетки умрут, а нормальные клетки восстановятся. Но поскольку он уничтожает все, наступает период, когда у пациента мало здоровых кровяных телец и возникает риск осложнений. С другой стороны, таргетная терапия атакует только раковые клетки, экономя все остальное и ограничивая риск осложнений.В настоящее время для лечения лейкозов используются несколько целевых препаратов по отдельности или в сочетании с химиотерапией, но их количество не за горами.
К сожалению, с хроническим миелоидным лейкозом никто не ждет. Но лечение — это таблетки. Исторически сложилось так, что пациенты принимали таблетки всю оставшуюся жизнь, но новые исследования показывают, что если пациент чувствует себя хорошо через пять или шесть лет, мы можем прекратить химиотерапию с низким риском рецидива рака.
Есть ли еще что-нибудь о лечении лейкемии, которым вы хотели бы поделиться?
Я очень взволнован, потому что думаю, что мы вылечим лейкоз еще при моей жизни.Мы уже зашли так далеко. Например, до 2000 года выживаемость пациентов с острым лимфобластным лейкозом с положительной филадельфийской хромосомой составляла 10%. Таргетная терапия улучшила этот показатель до 35-40%, но пациенты должны были пройти трансплантацию стволовых клеток, что было непросто. Но я руководил исследованием препарата под названием понатиниб в сочетании с химиотерапией, и мы наблюдали, как выживаемость подскочила до 80% без трансплантата. Наша следующая цель — увеличить выживаемость до 90% и исключить необходимость в химиотерапии, используя комбинацию таргетной терапии и иммунотерапии.
Я горжусь тем, что доктор медицины Андерсон лидирует. Здесь мы разработали понатиниб, и теперь это международный стандарт. Мы являемся первопроходцами в лечении этих пациентов без химиотерапии.
Запишитесь на прием в MD Anderson онлайн или позвонив по телефону 1-877-632-6789.
Острый миелоидный лейкоз (ОМЛ) | Онкологический совет штата Новый Южный Уэльс
Острый миелоидный лейкоз (ОМЛ) — это рак крови, который появляется внезапно и быстро растет. Это начинается, когда незрелые лейкоциты, называемые бластами, становятся злокачественными.Эти аномальные бластные клетки известны как лейкозные клетки. Они быстро размножаются и продолжают делиться, но никогда не созревают в нормальные клетки.
Поскольку лейкозные клетки незрелые и аномальные, они не выполняют обычную роль лейкоцитов в борьбе с инфекциями. Они также вытесняют нормальные лейкоциты, которые затем не могут работать должным образом. Это увеличивает риск заражения. Когда костный мозг наполняется лейкозными клетками, остается мало места для нормальных эритроцитов и тромбоцитов.Это может вызвать усталость, проблемы с кровотечением и другие проблемы со здоровьем.
Подробнее о:
В чем разница между острым и хроническим лейкозом?
Хотя все типы лейкемии начинаются в костном мозге и влияют на выработку лейкоцитов, они сгруппированы в зависимости от того, какой тип лейкоцитов поражен, есть ли аномалии в костном мозге и насколько быстро развивается болезнь.
Острый лейкоз обычно поражает неразвитые клетки, возникает внезапно и быстро растет.Хронический лейкоз обычно поражает частично развитые клетки, появляется постепенно и медленно растет в течение месяцев или лет. Для получения дополнительной информации см. Хронический лейкоз — ХЛЛ или Хронический лейкоз ХМЛ.
Есть два типа острого лейкоза:
В чем разница между AML и ALL?
AML и ALL — два основных типа острого лейкоза. Разница между ними заключается в типе пораженных лейкоцитов.
Острый миелоидный лейкоз (ОМЛ) — развивается из миелоидных клеток.Существуют различные подтипы AML, включая подтип, известный как острый промиелоцитарный лейкоз (APML), который развивается из незрелых миелоидных клеток, называемых промиелоцитами. APML лечится иначе.
Острый лимфобластный лейкоз (ОЛЛ) — ОЛЛ может развиваться из разных типов лимфоцитов, включая В-клетки или Т-клетки. Узнайте больше о том, как диагностировать и лечить ВСЕ.
Как возникает лейкемия
Костный мозг производит три основных типа клеток крови: лейкоциты, эритроциты и тромбоциты.Лейкемия начинается, когда патологические лейкоциты становятся злокачественными и переполняют костный мозг, прежде чем попасть в кровоток. Без лечения они могут распространяться на лимфатические узлы и некоторые органы.
Дети с острым лейкозомЭта информация предназначена для взрослых с диагнозом «острый лейкоз». Поскольку у детей и взрослых с острым лейкозом одинаковые типы тестов, лечения и побочные эффекты, большая часть информации, представленной здесь, также применима к детям. Нет двух одинаковых случаев острого лейкоза, поэтому важно подробно обсудить случай вашего ребенка с его врачами.Узнайте больше о детях с острым лейкозом.
Что вызывает ПОД?
Точные причины острого лейкоза еще не выяснены. Известно, что факторы, повышающие вероятность развития болезни, включают:
- предыдущее лечение химиотерапией или лучевой терапией
- с определенными генетическими нарушениями, такими как синдром Дауна
- воздействие высоких уровней радиации (например, ядерная авария)
- воздействие некоторых химических веществ, таких как бензол
- курение
- ожирение
- конкретных заболеваний крови, таких как миелодисплазия (для AML)
- некоторых вирусов, таких как вирус Эпштейна-Барра (для ALL).
Кто получает ПОД?
Ежегодно в Австралии у более чем 3700 человек диагностируется форма лейкемии, и это самый распространенный тип рака, диагностируемый у людей моложе 24 лет. Около 1400 из этих случаев относятся к острому лейкозу, что составляет около 1,1% случаев рака. в Австралии, поэтому в целом острый лейкоз встречается редко.
Около 1100 человек ежегодно заболевают острым миелоидным лейкозом (ОМЛ). ОМЛ становится все более распространенным с возрастом и чаще возникает после 65 лет.
Кровь
Кровь циркулирует по вашему телу, чтобы обеспечить ваши ткани кислородом и питательными веществами, а также удалить продукты жизнедеятельности. Он состоит из клеток крови, содержащихся в прозрачной жидкости, называемой плазмой.
Все клетки крови живут ограниченное время и нуждаются в постоянной замене. Большинство из них производится в костном мозге, который представляет собой губчатую часть в центре костей.
Костный мозг содержит стволовые клетки крови. Это неспециализированные клетки, которые обычно превращаются в один из трех основных типов клеток крови: красные кровяные тельца, белые кровяные тельца или тромбоциты.Каждый тип кровяных клеток выполняет определенную работу.
Есть два семейства стволовых клеток крови:
- лимфоидные стволовые клетки — развиваются в белые кровяные тельца, называемые лимфоцитами, которые являются важной частью иммунной системы
- миелоидные стволовые клетки — развиваются в эритроциты, тромбоциты и все белые клетки, кроме лимфоцитов .
Когда миелоидные лейкоциты растут ненормально, это называется острым миелоидным лейкозом, а когда лимфоидные лейкоциты ненормально растут, это называется острым лимфобластным лейкозом.
Что такое лимфатическая система?
Лимфатическая система — это часть иммунной системы, которая защищает организм от болезней и инфекций.