У российских пациентов нашли мутации, которые вызывают синдром Луи-Бар
Редкое наследственное заболевание, при котором ухудшается состояние нервной и иммунной систем, называется атаксией-телеангиэктазией, или синдромом Луи-Бар. Оно встречается примерно у 1% населения. У больных нарушается походка, координация движений и происходит стойкое расширение кровеносных сосудов глаз и кожи. Синдром вызывают мутации в гене АТМ, который несет информацию о белке, распознающем повреждения ДНК и участвующем в процессе их исправления. Если мутация есть, клетка недостаточно хорошо справляется с восстановлением своего генетического материала, а значит, увеличивается риск развития опухолей.
Ген АТМ большой, из-за этого проводить ДНК-диагностику сложнее. В недавней работе российские ученые и медики использовали новые технологии исследования мутаций, которые могут облегчить диагностику этого тяжелого заболевания. В течение двух лет в Москве и Санкт-Петербурге обследовали 17 детей с теми или иными клиническими признаками болезни: во всех случаях диагноз был подтвержден молекулярными методами.
У пациентов обнаружили три часто повторяющихся патогенных варианта гена. Исследователи отмечают, что две из трех обнаруженных мутаций гена характерны для пациентов из стран Восточной Европы, в частности Польши, России, Украины и Беларуси. Одну из этих мутаций впервые обнаружили у североамериканских меннонитов, которые проживали в России до конца XIX века. Третья разновидность ранее не была описана у славян, и ее географическое и этническое распространение еще предстоит уточнить. Женщины — обладательницы наследственного дефекта одной копии гена АТМ подвержены повышенному риску развития рака груди, поэтому им может понадобиться профилактическое наблюдение.
«Результаты нашей работы указывают на то, что более половины мутаций в гене АТМ у российских пациентов представлены всего тремя вариантами. Эти данные должны быть приняты во внимание при проведении ДНК-диагностики атаксии-телеангиэктазии», — заключил один из авторов статьи Евгений Суспицын, доцент кафедры общей и молекулярной медицинской генетики Санкт-Петербургского государственного педиатрического медицинского университета.


Работа выполнена сотрудниками СПбГПМУ, НИИ онкологии имени Н. Н. Петрова, Детской городской больницы №1 (Санкт-Петербург), Российской детской клинической больницы, Российского национального исследовательского медицинского университета имени Н. И. Пирогова, Северо-Западного государственного медицинского университета имени И. И. Мечникова и Санкт-Петербургского государственного университета.
Врожденный иммунодефицит: синдром Луи-Бар | Интернет-издание «Новости медицины и фармации»
Синдром Луи-Бар (врожденной атаксии-телеангиэктазии — А-Т) — врожденное иммунодефицитное состояние с преимущественным поражением Т-звена иммунитета, характеризуется аномальным развитием эмбриональных закладок и, по-видимому, неправильным взаимодействием эктодермы и мезодермы. Синдром Луи-Бар — это генетическое заболевание, которое наследуется по аутосомно-рецессивному типу.
Иммунодефицит и хромосомная нестабильность являются маркерами А-Т ( Ataxia — Teteangiectasia Mutated ), что кодирует синтез одноименной киназы. Клетки пациентов с А-Т характеризуются повышенной чувствительностью к радиации, дефектами клеточного цикла, клинические же проявления и иммунологические нарушения имеют существенные различия, отмечаются повышенная частота развития злокачественных опухолей и спонтанная хромосомная нестабильность, хромосомные поломки, вовлекающие преимущественно 7-ю и 14-ю хромосомы [2, 3].
Известно, что клеточный цикл делится на 4 фазы: митоз (М) и синтез ДНК ( S ), разделенные двумя перерывами Gl и G 2. Последовательность клеточного цикла выглядит следующим образом: G 1 — S — G 2 — M . После воздействия ионизирующего излучения происходят двунитевые разрывы ДНК. Если происходит репапарация ДНК, то клеточный цикл восстанавливается, если нет — происходит гибель клетки путем апоптоза или развивается мутантный клон.
Клинические проявления А-Т могут существенно отличаться у разных больных. Прогрессирующая мозжечковая атаксия и телеэнгиэктазии присутствуют у всех, часто встречаются пятна «кофе с молоком» на коже. Склонность к инфекциям колеблется от очень выраженной до весьма умеренной. Очень высока частота развития злокачественных новообразований, преимущественно лимфоидной системы.

Клиническая характеристика. Заболевание начинается в раннем детстве и проявляется в первую очередь мозжечковой атаксией (100 %). Отмечаются качание головы и туловища, нарушение походки, интенционный тремор и хореоатетоз (90–100 %). Характерными изменениями глаз являются нарушение движения глазного яблока (80–90 %), нистагм (90–100 %) и косоглазие. В возрасте от 2 до 6 лет появляются телеангиэктазии на конъюнктиве и открытых участках тела, слизистой мягкого и твердого неба. Важным признаком синдрома являются хронические респираторные инфекции (синуситы и пневмонии, 60–80 %). Наблюдаются отставание в росте, пигментные пятна или участки депигментации на коже, склеродермия, гипотония мышц, гипорефлексия и дизартрия. У больных часто развиваются злокачественные новообразования, причем в 10–30 % поражается лимфоретикулярная система.
При патологоанатомическом исследовании обнаруживают аплазию или гипоплазию тимуса, уменьшение размеров лимфатических узлов и селезенки, признаки мозжечковой дегенерации, фиброзную дисплазию яичников.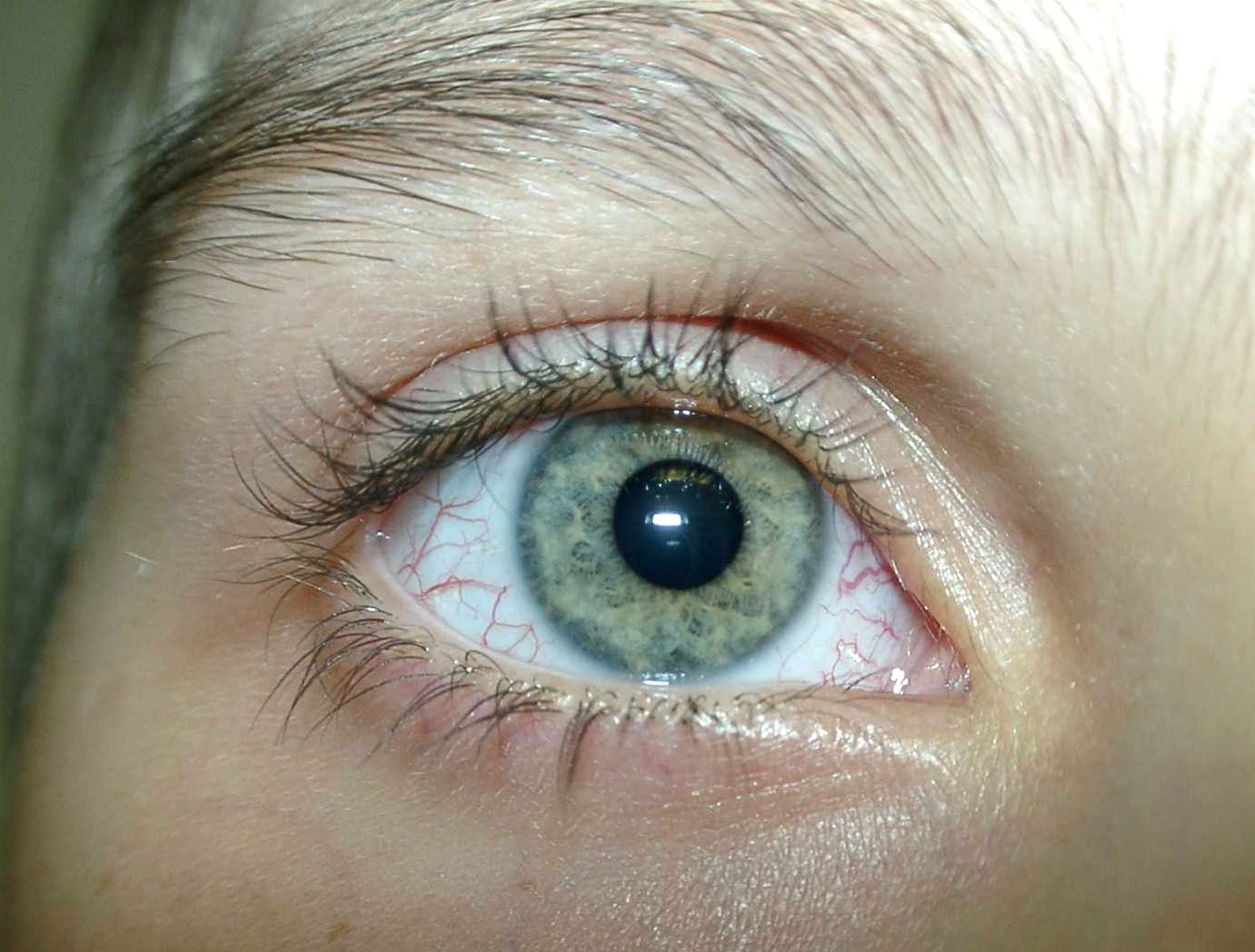 При А-Т имеет место нарушение В- и Т-клеточных систем иммунитета, которое выражается в отсутствии сывороточных иммуноглобулинов, главным образом IgA , но иногда IgG и IgE . При цитогенетическом исследовании лимфоцитов часто обнаруживают различные хромосомные аберрации и ломкость хромосом. Больные погибают от легочных инфекций либо от злокачественных новообразований [5].
При А-Т имеет место нарушение В- и Т-клеточных систем иммунитета, которое выражается в отсутствии сывороточных иммуноглобулинов, главным образом IgA , но иногда IgG и IgE . При цитогенетическом исследовании лимфоцитов часто обнаруживают различные хромосомные аберрации и ломкость хромосом. Больные погибают от легочных инфекций либо от злокачественных новообразований [5].
На первое место в клинической картине выступает неврологическая симптоматика, поэтому болезнь вначале была описана как мозжечковая атаксия. В возрасте от 2 до 8 лет возникают телеангиэктазии, которые обычно располагаются на бульбарной конъюнктиве, между углом глаза и лимбом, и имеют вид красных извитых сосудов. Наблюдается аплазия вилочковой железы, гипоплазия (недоразвитие) лимфатических узлов, селезенки, групповых лимфатических фолликулов тонкой кишки, миндалин. У детей с синдромом Луи-Бар постоянно наблюдается гипоплазия (недоразвитие) или аплазия (полное отсутствие) небных миндалин. Лакуны миндалин недоразвиты.
Диагноз ставится на основе клинической картины, а также данных лабораторных показателей. У всех больных с синдромом Луи-Бар почти полностью отсутствуют Т-супрессоры. У части больных клетки не могут синтезировать IgA, что связано с отсутствием Т-хелперов. В крови обнаруживают a- и b-протеин. Патогенетическим методом лечения является аллотрансплантация неонатальной вилочковой железы. Назначают курс инъекций активных факторов вилочковой железы (Т-активина, тималина, тимацина и др.), систематически вводят нативную плазму и нормальный иммуноглобулин человека [5].
Под нашим наблюдением находилась девочка К., в клинику она поступила в возрасте 13 лет и 10 месяцев по поводу врожденного иммуннодефицитного состояния с атаксией (синдром Луи-Бар), хронической пневмонии, полисегментарного пневмосклероза, гнойного деформирующего эндобронхита, бронхоэктатической болезни в фазе обострения, правосторонней крупноочаговой пневмонии, осложненных генерализованным амилоидозом внутренних органов: печени с развитием цирроза и печеночной недостаточности, почек, селезенки, кишечника, анемии, кахексии.
При поступлении жалобы матери на желтушное окрашивание кожи, повторную рвоту, анорексию, общую слабость, исхудание. Из анамнеза известно, что родилась доношенной, с малым весом — 2 700 г , с оценкой по шкале Апгар 6–7 баллов. Находилась на естественном вскармливании, до года не болела. Со второго года жизни отмечались частые простудные заболевания, стало прогрессировать исхудание, перенесла повторную пневмонию. С 4 лет выявлена мозжечковая атаксия. Девочка консультирована в нашей клинике, в клинике г. Москвы диагностирован синдром Луи-Бар. С тех пор прогрессировали явления дистрофизации, атаксия, переносила повторные пневмонии. Диагностирована хроническая бронхоэктатическая болезнь. Неоднократно лечилась в стационаре. Последние 2 года жизни девочка не ходит, присоединились изменения со стороны печени и почек, связанные с амилоидозом. За 3 месяца до последней госпитализации находилась в клинике, диагноз подтвержден, получала комплексную терапию — антибиотики широкого спектра действия, дезинтоксикационную терапию, иммунотерапию. Состояние девочки стабилизировалось. Выписалась домой на поддерживающей дозе препаратов, улучшающих обменные процессы печени и почек. За 2 недели до поступления состояние пациентки резко ухудшилось, наросла желтуха, наблюдалась полная анорексия, появилась повторная рвота. Направлена в клинику.
Состояние девочки стабилизировалось. Выписалась домой на поддерживающей дозе препаратов, улучшающих обменные процессы печени и почек. За 2 недели до поступления состояние пациентки резко ухудшилось, наросла желтуха, наблюдалась полная анорексия, появилась повторная рвота. Направлена в клинику.
При поступлении общее состояние тяжелое. Девочка резко дистрофизирована. Кожные покровы и склеры иктеричные, множественная «звездчатая» сыпь. На глазных яблоках выражен сосудистый рисунок. Заторможена, на вопросы отвечает вяло. Положение в постели горизонтальное, сидит с поддержкой. Видимые слизистые бледные. Язык розовый. Периферические лимфатические узлы мелкие, единичные до 0,5–1,0 см в диаметре, пальпируются подчелюстные. Пульс — 100. ЧД — 40. АД — 100/60 мм рт.ст. Над легкими перкуторно легочный звук, укорочен в нижних отделах, аускультативно дыхание жесткое, в нижних отделах ослабленное, выслушиваются единичные влажные мелкопузырчатые хрипы. Границы сердца расширены в поперечнике, левая — по передней аксиллярной линии. Тоны приглушены, ритмичны. Живот увеличен в объеме, при пальпации мягкий, асцита нет. Печень плотная, пальпируется на 4 см ниже реберной дуги, селезенка плотная, пальпируется на 5 см ниже реберной дуги у входа в малый таз. Мочится свободно. Стул оформлен, оправляется самостоятельно.
Тоны приглушены, ритмичны. Живот увеличен в объеме, при пальпации мягкий, асцита нет. Печень плотная, пальпируется на 4 см ниже реберной дуги, селезенка плотная, пальпируется на 5 см ниже реберной дуги у входа в малый таз. Мочится свободно. Стул оформлен, оправляется самостоятельно.
Анализ крови: Эр. — 2,9 Т/л, Н b — 90 г/л, Ц.П — 0,9, Лейк. — 8,2 Г/л, выражен анизоцитоз и пойкилоцитоз, п/я — 14 %, с/я — 20 %, л. — 64 %, м. — 2 %, СОЭ — 6 мм/час. Остаточный азот крови — 54,5 г/л. Холестерин крови — 4 мкмоль/л. АСТ — 0,35, АЛТ — 0,42. Общий билирубин крови — 84,8 ммоль/л, прямой — 74,2, непрямой — 10,6.
Сулемовая проба — 1,6. Общий белок крови — 64 г/л, альбумины — 46,7, гамма-глобулины — 19 %. Протромбин крови — 75 %.
Анализ мочи: белок — 0,86 г/л, Лейк. — 10–15, до 25 в п/зр., Эр. — 10 в п/зр., цилиндры гиалиновые — 1–2, зернистые — 1–2 в п/зр.
На рентгенограмме органов грудной клетки: легочная ткань умеренно вздута, особенно в нижних долях. Легочный рисунок усилен, расширен, справа в средней доле крупноочаговая инфильтрация легочной ткани без четких контуров. Синусы свободны. Сердце — в норме. ЭКГ: диффузное поражение миокарда. На основании анамнеза, объективных данных, клинического обследования и наблюдения выставлен вышеописанный диагноз.
Легочный рисунок усилен, расширен, справа в средней доле крупноочаговая инфильтрация легочной ткани без четких контуров. Синусы свободны. Сердце — в норме. ЭКГ: диффузное поражение миокарда. На основании анамнеза, объективных данных, клинического обследования и наблюдения выставлен вышеописанный диагноз.
Получала терапию: в/в капельно р-р Рингера, гемодез, плазма, корглюкон, лазикс, ампициллин в/м, ежедневно гамма-глобулин, сирепар, липоевая кислота, метионин, преднизолон, оксигенотерапия, диета № 7.
Несмотря на проводимую терапию, состояние девочки прогрессивно ухудшалось, нарастали явления печеночной и почечной недостаточности, уменьшался суточные диурез, последние дни до 300 г в сутки. В легких увеличилось количество хрипов, нарастала дыхательная и сердечная недостаточность. Через 18 дней после поступления в стационар состояние агонирующее, появилось носовое кровотечение, в кале примесь крови, стул дегтеобразный, появился печеночный запах. Проводимые реанимационные мероприятия эффекта не дали. При явлении печеночной с присоединением дыхательной и сердечной недостаточности девочка умерла на 20-й день пребывания в клинике.
При явлении печеночной с присоединением дыхательной и сердечной недостаточности девочка умерла на 20-й день пребывания в клинике.
Основной : врожденное иммунодефицитное состояние с атаксией — синдром Луи-Бар. Хроническая пневмония. Полисегментарный пневмосклероз, гнойный деформирующий эндобронхит, бронхоэктатическая болезнь в стадии обострения, правосторонняя крупноочаговая пневмония.
Осложнения: генерализованный амилоидоз внутренних органов: печени с развитием цирроза и печеночной недостаточности, почек, селезенки, кишечника. Анемия. Кахексия.
Особенностью данного клинического случая можно считать редкую частоту встречаемости, характерную клиническую и лабораторную картину заболевания, медленное прогрессирование развития синдрома Луи-Бар, возраст пациентки.
Луи-Бар синдром
Луи-Бар синдром (синонимы: атаксия-телеангиэктазия, цефало-окуолокутанная телеангиэктазия. )
)
В 1941 г. Луи-Бар описала синдром, характеризующийся мозжечковой атаксией и кожно-конъюнктивальной телеангиэктазией. В 1958 г. Boder и Sedgwick предложили название атаксия-телеангиэктазия и добавили третий важный компонент этого синдрома — рецидивирующую тяжелую синопульмональную инфекцию.
Этиология и патогенез
Этот синдром в различных классификациях рассматривается как спинно-мозжечковая дегенерация (Toller и Millichap) или как факоматоз (Boder, Miller и др.). Термин факоматоз (phakomatosrls) предложил van der Hoeve в 1923 г. для заболеваний с комбинированным поражением нервной системы и кожи (врожденные нейро-экто-мезодермальные дисплазии) Boder и Sedgwick в 150 случаях синдрома Луи-Бар обнаружили дегенеративные явления в ЦНС, дегенерацию пирамидальных клеток, расширение менингеальных сосудов и др.
Значительная часть описанных наблюдений относится к семейно-наследственной патологии, тип наследования аутосомно-рецессивный.
Многие современные дерматологи синдром Луи-Бар относят к сочетанным иммунодефицитным состояниям. В основе лежат гипоплазия тимуса и дефицит IgA и IgE, т. е. нарушение функции клеточного и гуморального звеньев иммунитета, чем и объясняют частые рецидивирующие инфекционные заболевания органов дыхания, пищеварительного тракта, кожи и др. Помимо гипоплазии вилочковой железы отмечается гипо- атрофия лимфатических узлов, селезенки и лимфатического аппарата, пищеварительного канала. Снижение иммунитета приводит к ранней гибели больных от инфекционных заболеваний и усиливает тенденцию к развитию злокачественных новообразований лимфатической системы.
В основе лежат гипоплазия тимуса и дефицит IgA и IgE, т. е. нарушение функции клеточного и гуморального звеньев иммунитета, чем и объясняют частые рецидивирующие инфекционные заболевания органов дыхания, пищеварительного тракта, кожи и др. Помимо гипоплазии вилочковой железы отмечается гипо- атрофия лимфатических узлов, селезенки и лимфатического аппарата, пищеварительного канала. Снижение иммунитета приводит к ранней гибели больных от инфекционных заболеваний и усиливает тенденцию к развитию злокачественных новообразований лимфатической системы.
Клиника
Заболевание встречается редко. Оно включает в себя поражения нервной системы, кожно-конъюнктивальные телеангиэктазии и инфекционные заболевания. Среди неврологических манифестаций этого синдрома атаксия мозжечкового характера является первым симптомом у всех больных; начинается с раннего детства, становится особенно заметной, когда ребенок начинает ходить. Вследствие прогрессирования мозжечковых расстройств больные часто полностью лишены возможности ходить. Отмечается атаксия позы и конечностей, гипотония мышц, замедленная скандированная невнятная речь по типу мозжечковой дизартрии, нистагм, тремор, нарушения иннервации движений глазных яблок. Сухожильные рефлексы низкие или отсутствуют. Окуломоторная апрексия встречается в 80% случаев, затем развивается офтальмоплегия.
Отмечается атаксия позы и конечностей, гипотония мышц, замедленная скандированная невнятная речь по типу мозжечковой дизартрии, нистагм, тремор, нарушения иннервации движений глазных яблок. Сухожильные рефлексы низкие или отсутствуют. Окуломоторная апрексия встречается в 80% случаев, затем развивается офтальмоплегия.
Телеангиэктазии обычно появляются после атаксии, в возрасте 4-6 лет, в некоторых случаях позже, а иногда на первом месяце жизни. Телеангиэктазии вначале развиваются на глазных яблоках (бульбарная конъюнктива), затем на веках и коже лица.
Инфекционные заболевания главным образом органов дыхания обнаруживаются у 80% больных. Они проявляются рецидивирующими рино-фарингитами, хроническими бронхитами, бронхопневмониями, бронхоэктазиями. Описываются гнойные отиты. Патогенетически их связывают с иммунным дефицитом.
Дерматологическая картина синдрома Луи-Бар характеризуется наличием кожных телеангиэктазий у 100% больных. Другие кожные проявления (резкая сухость кожи и волос, фолликулярный кератоз на предплечьях и голенях, пигментации цвета «кофе с молоком» и сетчатые дисхроматически-атрофические поражения на коже лица) наблюдаются у 50-70% больных.
Кожные изменения не являются специфическими для атаксии — телеангиэктазии, однако диагностическая важность этих изменений делает необходимым их раннее выявление в целях более успешного лечения; часто диагноз ставится на основании дерматологической картины.
Вначале телеангиэктазии появляются на бульбарной конъюнктиве, затем распространяются на кожу век, носа, ушных раковин, шеи; появляются на коже локтевых сгибов, предплечий, подколенных областей, тыльной поверхности кистей, стоп. Телеангиэктазии особенно выражены наг участках кожи, которые подвергаются солнечному облучению. На лице, в области щек, они принимают вид «пучков» и расширения сосудистой сети. Веки часто становятся неэластичными, кожа лица плотной, напоминающей склеродермию.
Иногда телеангиэктазии также наблюдаются на мягком и твердом нёбе. Из других кожных проявлений болезни, имеющих важное значение для диагноза, следует отметить гиперпигментацию и гипопигментацию с атрофией и телеангиэктазиями, напоминающие пойкилодермию.
Луи-Бар в оригинальном описании указывала на наличие пятен цвета кофе с молоком, размерами от чечевицы и больше, напоминающих веснушки на коже носа и щек, подбородка, груди и межлопаточной области. Кроме того, у многих больных наблюдаются фолликулярный кератоз и сухость кожи. У части больных наблюдаются экзематозные поражения, преждевременное поседение волос, гипертрихоз, псориазоподобные высыпания, акнеиформные элементы на щеках и др.
Прогноз неблагоприятный, более половины больных погибают через 5-10 лет после появления первых симптомов болезни от воспаления легких или лимфоретикулярных злокачественных опухолей.
Лечение
В связи с частыми инфекционными заболеваниями легких необходимо назначение антибиотиков, сульфаниламидов и других противовоспалительных препаратов. Больные подлежат диспансерному наблюдению у невролога, педиатра и дерматолога.
Клинический случай синдрома Луи-Бар Текст научной статьи по специальности «Клиническая медицина»
КЛИНИЧЕСКИЙ СЛУЧАЙ СИНДРОМА ЛУИ-БАР
№3-2015
Г. Б. КАДРЖАНОВА, А.Р. СМАГУЛОВА, Г.А. МУХАМБЕТОВА, К.С. САРБАСОВА
Б. КАДРЖАНОВА, А.Р. СМАГУЛОВА, Г.А. МУХАМБЕТОВА, К.С. САРБАСОВА
Казахский Национальный медицинский университет имениС. Д. Асфендиярова, кафедра нервных болезней. Университетская клиника «Аксай» г. Алматы, Казахстан
УДК 616.8-056.76
В статье приведен случай собственного клинического наблюдения синдрома Луи-Бар (врожденной атаксии-телеангиэктазии) у мальчика 14 лет. Отмечены особенности течения, клинические признаки и дополнительные методы исследования синдрома Луи-Бар.
Ключевые слова: Синдром Луи-Бар, атаксия, телеангиэктазия.
Синдром Луи-Бар_ впервые описан в 1941 году современным французским врачом D. Louis Bar, позднее — американскими врачами E. Boder, R. P. Sedgwick; синоним — атаксия телеангиэктазия. Это — наследственное заболевание, начинающееся в раннем детском возрасте с неврологической симптоматикой, снижением
иммунологической реактивности организма, относится к группе факоматозов. Атаксия-телеангиэктазия — заболевание с аутосомно-рецессивным типом наследования, характеризующееся нарушением репарации ДНК, тяжелым иммунодефицитом, мозжечковой дегенерацией,
Атаксия-телеангиэктазия — заболевание с аутосомно-рецессивным типом наследования, характеризующееся нарушением репарации ДНК, тяжелым иммунодефицитом, мозжечковой дегенерацией,
телеангиэктазиями различной локализации,
предрасположенностью к онкологическим заболеваниям, сенситивностью к радиационным воздействиям. Частота возникновения 1:40 000 живорожденных. Ген локализован на хромосоме 11 (q22-23). При обследовании отмечаются мозжечковая атаксия, которая обычно становится заметной для окружающих в период приобретения ребенком навыков ходьбы, дизартрия, глазодвигательные нарушения, хореоатетоз, миоклонии, а также эндокринные нарушения. Характерные кожные изменения (телеангиэктазии) появляются в 3—6 лет. Первичная локализация телеангиэктазии — конъюнктива глаз, с последующим распространением на лицо, шею, небо, ушные раковины, дорсальную поверхность рук [1].
Заболевание начинается в раннем детстве, проявляется в первую очередь мозжечковой атаксией (100%). Отмечаются качание головы и туловища, нарушение походки, интенционный тремор и хореоатетоз (90-100%), характерным изменением глаз являются нарушения движения глазных яблока (80-90%), нистагм 99-100%) и косоглазие [2]. При этой редкой форме факоматоза наблюдаются неврологические симптомы, кожные проявления в виде паукообразного разрастания сосудов (телеангиэктазии), снижение иммунологической реактивности организма. Первые признаки болезни появляются в возрасте одного года — четырех лет. Походка становится неустойчивой, появляется неловкость движений, нарушение плавности речи (скандированная речь). Прогрессирование мозжечковых нарушений постепенно приводит к тому, что больные перестают ходить. Дети с атаксией-телеангиэктазией часто болеют простудными заболеваниями, воспалением придаточных пазух носа, воспалением легких. Они обусловлены снижением защитных иммунологических свойств крови, отсутствием специфических антител на фоне прогрессирования болезни нарастают нарушения интеллекта, расстраиваются внимание, память, снижается способность к абстрации.
Отмечаются качание головы и туловища, нарушение походки, интенционный тремор и хореоатетоз (90-100%), характерным изменением глаз являются нарушения движения глазных яблока (80-90%), нистагм 99-100%) и косоглазие [2]. При этой редкой форме факоматоза наблюдаются неврологические симптомы, кожные проявления в виде паукообразного разрастания сосудов (телеангиэктазии), снижение иммунологической реактивности организма. Первые признаки болезни появляются в возрасте одного года — четырех лет. Походка становится неустойчивой, появляется неловкость движений, нарушение плавности речи (скандированная речь). Прогрессирование мозжечковых нарушений постепенно приводит к тому, что больные перестают ходить. Дети с атаксией-телеангиэктазией часто болеют простудными заболеваниями, воспалением придаточных пазух носа, воспалением легких. Они обусловлены снижением защитных иммунологических свойств крови, отсутствием специфических антител на фоне прогрессирования болезни нарастают нарушения интеллекта, расстраиваются внимание, память, снижается способность к абстрации. Дети быстро истощаются. Отмечается резкие изменения настроения [3]. Постепенно развиваются атрофии кожи, поседение волоса уже в школьном возрасте, задержка психического и физического развития. Обычны гипоплазия мозжечка, резче выраженная в его черве, гипоплазия вилочковой железы, дисгаммоглобулинемия, поражение мононуклеарных
Дети быстро истощаются. Отмечается резкие изменения настроения [3]. Постепенно развиваются атрофии кожи, поседение волоса уже в школьном возрасте, задержка психического и физического развития. Обычны гипоплазия мозжечка, резче выраженная в его черве, гипоплазия вилочковой железы, дисгаммоглобулинемия, поражение мононуклеарных
макрофагов (ретикулезы, лимфосаркомы и др.). Прогноз плохой. Причиной смерти чаще являются хронические заболевания бронхов и легких, лимфомы, карциномы [4]. Нейрорадиологические исследования выявляют
прогрессирующую атрофию мозжечка, преимущественно червя, вторичную атрофическую дилатацию IV желудочка, постгеморрагические очаги в паренхиме мозга, появление которых связано с кровоизлияниями из эктазированных сосудов. Наличие у ребенка атрофических изменений мозжечка, визуализируемых на КТ, МРТ головного мозга, в сочетании с атаксией и прогрессирующим неврологическим дефицитом требует исключения синдрома атаксии-телеангиэктазии [1]. Собственное наблюдение.
Собственное наблюдение.
Под нашим наблюдением находился мальчик Х., 2000 года рождения, в клинику он поступил в возрасте 14 лет, верифицирован клинический диагноз «синдром Луи-Бар». При поступлении жалобы матери на шаткость при ходьбе, плохо разговаривает, задержку в психическом развитии, общую слабость, часто болеет простудными заболеваниями, частый влажный кашель. Из анамнеза жизни известно, что мальчик родился от 6 беременности, 6 родов. Беременность протекала без особенностей, родился в срок. Вес при рождении 3200,0, закричал сразу, длина тела 50 см. Самостоятельно мальчик стал ходить с 1,5 лет, развивается с задержкой в речевом развитии. Анамнез заболевания: со слов матери мальчик болен с 5 лет, когда впервые обратили внимание, что ребенок стал хуже ходить. В 6 лет пошел в школу, но учиться не смог, с детьми не общался, со школьной программой не справлялся, школу посещал только в течение 1-го месяца. Затем обучался на дому, учебными навыками ребенок не овладел, не научился читать, писать, буквы не знает, при написании букв отмечалось дрожание правой руки. Со временем постепенно нарастала общая слабость, изменилась походка, стал шататься при ходьбе, часто падать, быстро уставать, меньше разговаривать. В 6 летнем возрасте появилось покраснение глаз, телеангиэктазии. Приступов судорог у мальчика не было. С 7 лет появился интенционный тремор в руках. В ноябре 2008 года установлен диагноз: Атаксия телеангиоэктазия. Синдром Луи-Бар. Мальчик неоднократно находился на стационарном лечении в неврологическом отделении по месту жительства. Наследственность отягощена: в семье 1 девочка умерла в возрасте 13 лет — у девочки с 8 лет прогрессивно ухудшалось состояние, нарастала слабость, появилась шаткость при ходьбе, затем тремор рук. С 8 лет перестала ходить, часто болела простудными заболеваниями. Диагноз у девочки не был установлен, симптомы заболевания были аналогичны, как у данного ребенка. В семье еще 3 девочки здоровы (26 лет, 24 года, 17 лет), мальчик 11 лет — здоров. 1 мальчик умер в 8 месяцев от кишечной инфекции. Брак близкородственный (родители — двоюродные брат и сестра), по национальности дунгане.
Со временем постепенно нарастала общая слабость, изменилась походка, стал шататься при ходьбе, часто падать, быстро уставать, меньше разговаривать. В 6 летнем возрасте появилось покраснение глаз, телеангиэктазии. Приступов судорог у мальчика не было. С 7 лет появился интенционный тремор в руках. В ноябре 2008 года установлен диагноз: Атаксия телеангиоэктазия. Синдром Луи-Бар. Мальчик неоднократно находился на стационарном лечении в неврологическом отделении по месту жительства. Наследственность отягощена: в семье 1 девочка умерла в возрасте 13 лет — у девочки с 8 лет прогрессивно ухудшалось состояние, нарастала слабость, появилась шаткость при ходьбе, затем тремор рук. С 8 лет перестала ходить, часто болела простудными заболеваниями. Диагноз у девочки не был установлен, симптомы заболевания были аналогичны, как у данного ребенка. В семье еще 3 девочки здоровы (26 лет, 24 года, 17 лет), мальчик 11 лет — здоров. 1 мальчик умер в 8 месяцев от кишечной инфекции. Брак близкородственный (родители — двоюродные брат и сестра), по национальности дунгане. У родителей мальчика 4 внука
У родителей мальчика 4 внука
№3-2015
от старших дочерей — все здоровы. Брак у детей неродственный.
Состояние мальчика тяжелое по поражению центральной нервной системы. В контакт мальчик вступает замедленно, формально. Поведение пассивное, настроение депрессивное, обращенную речь в пределах быта понимает. Сознание ясное. Когнитивные функции снижены: внимание неустойчивое, память снижена, мышление торпидное, бедная мимика. Навыками чтения и письма не владеет, математические представления и навыки развиты слабо. Запас знаний, представлений ниже возрастной нормы. Темп
речи замедлен, в активе — короткая фраза из 2-3 слов, словарный запас значительно ограничен,
звукопроизношение нарушено. Фонематический слух развит слабо.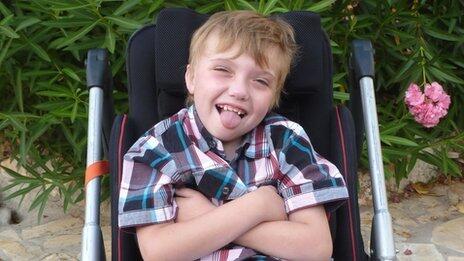 Состояние артикуляционного аппарата: ограничена подвижность языка и губ. Окружность головы 49 см (-5 см), микроцефальной формы. Черепно-мозговая иннервация -лицо симметричное, движения глазных яблок в полном объеме, горизонтальный нистагм. У мальчика выражены проявления мозжечковой атаксии: ходит при поддержке, походка шаткая с широко расставленными ногами, мегалография (рисунок 1).
Состояние артикуляционного аппарата: ограничена подвижность языка и губ. Окружность головы 49 см (-5 см), микроцефальной формы. Черепно-мозговая иннервация -лицо симметричное, движения глазных яблок в полном объеме, горизонтальный нистагм. У мальчика выражены проявления мозжечковой атаксии: ходит при поддержке, походка шаткая с широко расставленными ногами, мегалография (рисунок 1).
Рисунок 1
В позе Ромберга — статическая атаксия. При выполнении пальце-носовой и пяточно-коленной проб отмечается дисметрия, интенционный тремор; положительные феномены Стюарт-Холмса и асинергия Бабинского с 2-х сторон. Мышечный тонус диффузно снижен. Сухожильные рефлексы с рук и ног не вызываются. Имеются симптомы поражения экстрапирамидной системы в виде гипокинезии,
гипомимии, брадилалии, брадипсихии. Мальчик астенического телосложения, пониженного питания, подкожно-жировой слой истончен. Ребенок часто болеет респираторными инфекциями, отстает в физическом развитии от своего возраста в росте и весе: вес 18 кг, рост 130 см. С 2-х сторон отмечаются в области конъюнктивы телеангиэктазии (рисунки 2,3).
Ребенок часто болеет респираторными инфекциями, отстает в физическом развитии от своего возраста в росте и весе: вес 18 кг, рост 130 см. С 2-х сторон отмечаются в области конъюнктивы телеангиэктазии (рисунки 2,3).
Рисунок 2
Рисунок 3
На коже лица и ушных раковин отмечаются пигментные пятна, веснушки на лице, сухость кожи. Видимые слизистые бледные, язык розовый. Периферические лимфатические узлы мелкие. Пульс — 86 в 1 минуту, частота дыхания— 24 в 1 минуту, артериальное давление — 90/60 мм рт.ст. Над легкими перкуторно легочный звук, аускультативно дыхание жесткое, выслушиваются влажные проводные хрипы. Сердечные тоны умеренно приглушены, ритмичны. Живот при пальпации мягкий, безболезненный. Печень и селезенка не увеличены. Стул и диурез не нарушены. Проведено клиническое и лабораторное обследование: Общий анализ кров от 28.04. 2015 г: эритроцита 4,52*1012/л, гемоглобин 146 г/л, цветной показатель 0,96, лейкоциты 6,2*10*109/л, сегментоядерные 50, эозинофилы 5, лимфоциты 10, палочкоядерные 35. СОЭ 10 мм/час. (лимфоцитопения). Общий анализ мочи от 28.04.2015 г.:
2015 г: эритроцита 4,52*1012/л, гемоглобин 146 г/л, цветной показатель 0,96, лейкоциты 6,2*10*109/л, сегментоядерные 50, эозинофилы 5, лимфоциты 10, палочкоядерные 35. СОЭ 10 мм/час. (лимфоцитопения). Общий анализ мочи от 28.04.2015 г.:
количество 50 мл, удельный вес 1030, реакция — кислая, РН 6.0, лейкоциты 2-3 в поле зрения. Психолог: Интеллектуальное развитие ниже возрастной нормы. Логопед: Общее недоразвитие речи II уровень. Мозжечковая дизартрия. Компьютерная томография головного мозга от 29.04.2015 г.: на серии КТ сканов в MPR реконструкциях негрубо расширены экстрацеребральные ликворные пространства. Срединные структуры не смещены. Умеренно расширены боковые желудочки. Очаговых изменений в веществе головного мозга не выявлено. В задней черепной ямке расширены субарахноидальные пространства. В намете мозжечка: ликворные образования. Миндалины мозжечка гипоплазированы. Заключение: энцефалопатия с атрофическими изменениями в мозжечке с наличием арахноидальной кисты в намете мозжечка (рисунок 4).
Са*
№3-2015
Рисунок 4
Рентгенологическое исследование органов грудной клетки от 27.04.2015 г.: на рентгенограмме грудной клетки форма ее изменена: бочкообразная с расширением межреберных промежутков в верхних отделах. Очаговые и инфильтративные тени в легких не определяются. Легочный рисунок деформирован, сгущен в медиальных
долях. Корни легких расширены, бесструктурные. Сердце вертикально расположено «капельное», уменьшено в размерах, синусы свободны. Заключение: картина хронического деформирующего бронхита с эмфиземой в стадии обострения (рисунок 5).
Окулист от 28.04.2015 г.: диски зрительных нервов розовые монотонно окрашены, округлые, границы четкие. Вены нормального калибра, извиты, артерии ссужены, умеренно извиты. На конъюнктиве с 2-х сторон телеангиэктазии в виде паукообразного разрастания сосудов. DS Ои Телеангиэктазии. Ангиопатия сетчатки. Выводы. Особенностью данного клинического случая явилось то, что ребенок родился от близкородственного брака с отягощенной наследственностью. Синдром Луи-Бар является аутосомно-рецессивным заболеванием, т. е. проявляется клинически только при получении
Вены нормального калибра, извиты, артерии ссужены, умеренно извиты. На конъюнктиве с 2-х сторон телеангиэктазии в виде паукообразного разрастания сосудов. DS Ои Телеангиэктазии. Ангиопатия сетчатки. Выводы. Особенностью данного клинического случая явилось то, что ребенок родился от близкородственного брака с отягощенной наследственностью. Синдром Луи-Бар является аутосомно-рецессивным заболеванием, т. е. проявляется клинически только при получении
к 5
рецессивного гена сразу от обоих родителей. Синдром Луи-Бар это редкое заболевание. В связи со снижением иммунологической реактивности организма пациенты с синдром Луи-Бар должны проходить лечение и наблюдение не только у невролога, но и у иммунолога, отоларинголога, офтальмолога, пульмонолога, эндокринолога, онколога. Детям с синдромом Луи-Бар необходимо проведение иммунограммы с определением Т-супрессоров. С профилактической целью рождения детей с синдромом Луи-Бар рекомендовано братьям и сестрам мальчика не вступать в родственные браки.
СПИСОК ЛИТЕРАТУРЫ
1 А. С. Петрухин. Детская неврология. — М.: 2012. — Т 2. — 560 с.
2 С. И. Козлова. Е. Семанова, Н. С. Денисова, О. Е. Блинникова. Наследственные синдромы и медико-генетическое консультирование. Справочник. — М.: Медицина, 1987. — 320 с.
3 Л. О Бадалян. Детская неврология. — 3-е изд. — М.: Медицина, 1984, — 576 с.
4 Гусев Е. И., Бурд Г. С., Никифоров А.Неврологические симптомы, синдромы, симптомокоплескы и болезни. — М.: Медицина, 1999. — 880 с.
Г. Б. КАДРЖАНОВА, А. Р. СМАГУЛОВА, Г.А. МУХАМБЕТОВА, К. С. САРБАСОВА
ЛУИ-БАР СИНДРОМЫНЫН, КЛИНИКАЛЫК ЖАF ДАЙЫ
ТYЙiн: Ма;алада Луи-Бар синдромымен (туа пайда болган атаксия-телеангиэктазия)14 жастагы баланыц жеке ба;ылаудагы клиникальщ жагдайы керсетшген.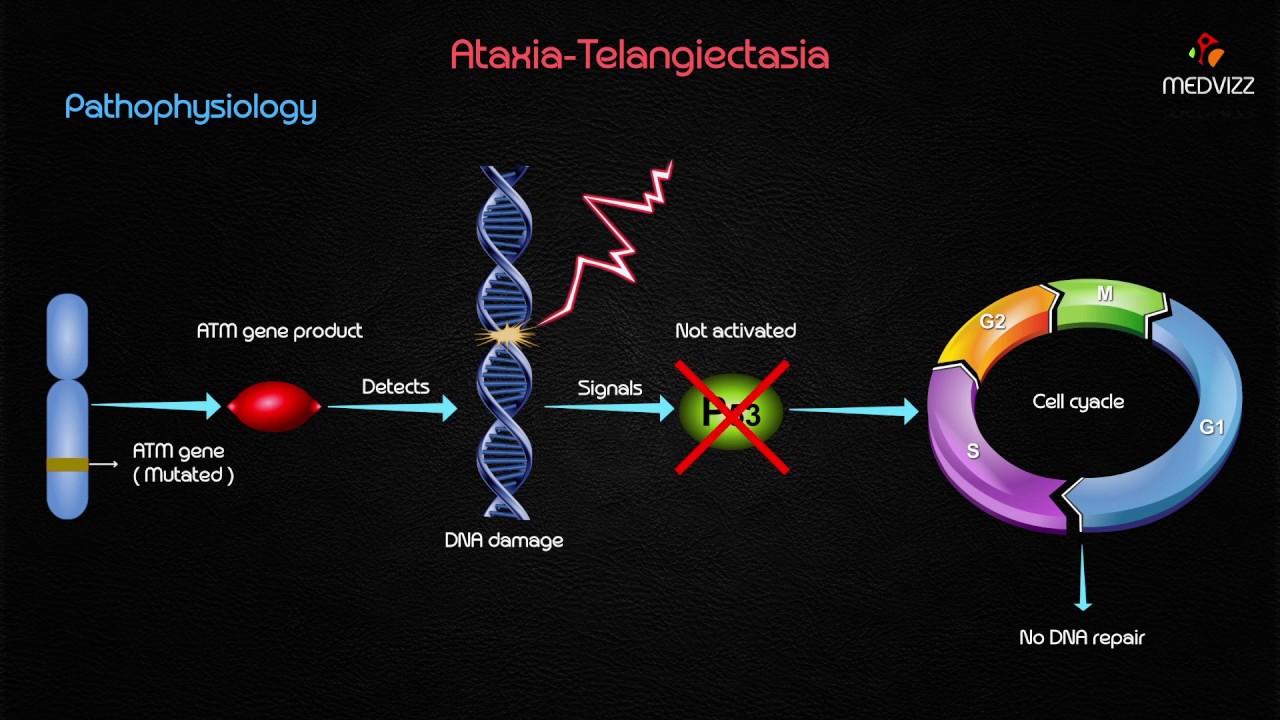 Каз®|1У
Каз®|1У
№3-2015
G.KADRZHANOVA, A. SMAGULOVA, G. MUCHAMBETOVA, K. SARBASOVA
A CLINICAL CASE OF LOUIS-BAR SYNDROME
Resume: The article describes the case of own clinical observations of Louis-Bar syndrome (congenital ataxia-telangiectasia) the boy is 14 years old. Marked features of the course, clinical signs and additional methods of research of Louis-Bar syndrome. Keywords: Louis-Bar syndrome, ataxia, elangiectasia
БОЛЕЗНЬ АЛЬЦГЕЙМЕРА -СЛЕДСТВИЕ ПОРАЖЕНИЯ СОСУДОВ ГОЛОВНОГО МОЗГА
М.Т. МЕРГЕНБАЕВА, А.А. НУРМУХАНБЕТОВА
Казахский Национальный медицинский университет им.С.Д.Асфендиярова
УДК 616.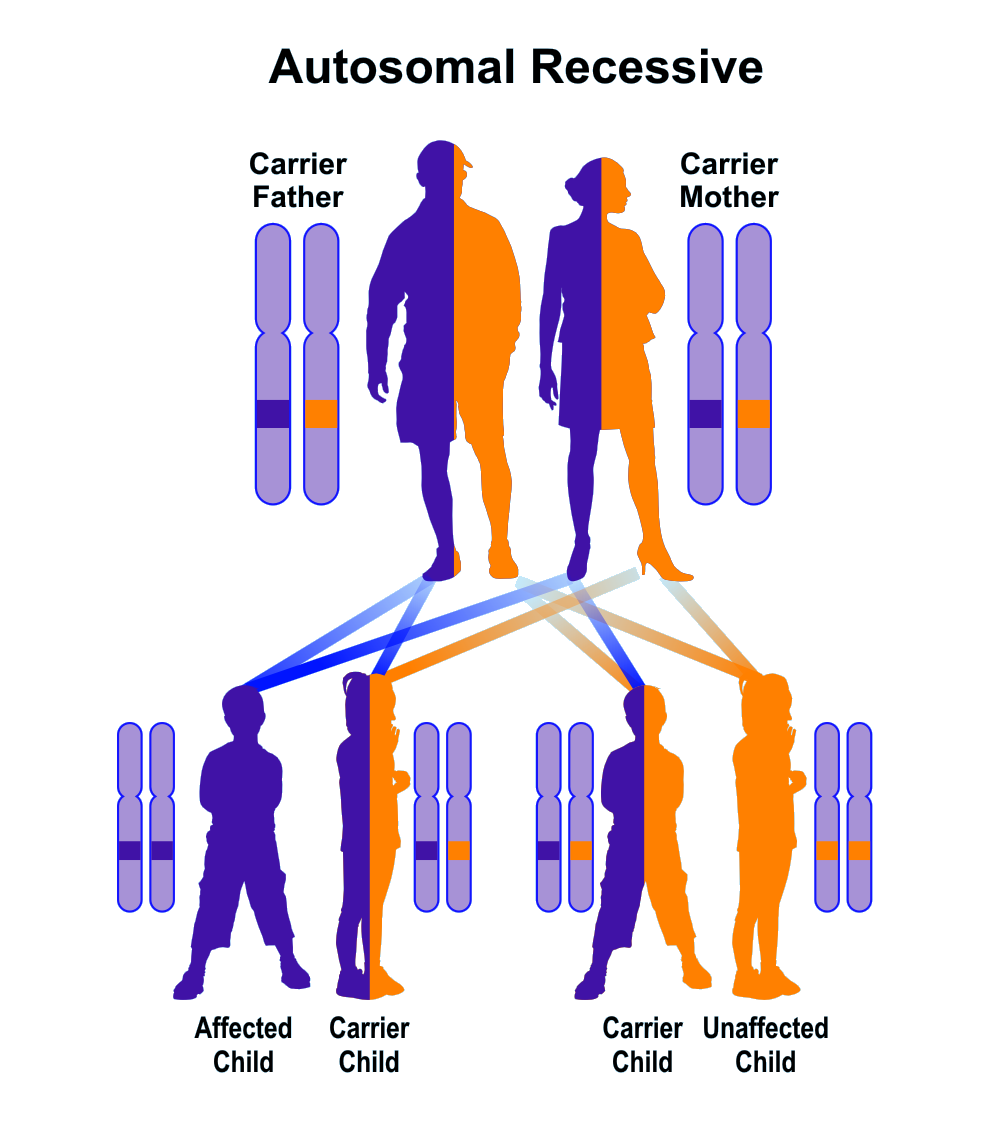 892.32:616.831-005
892.32:616.831-005
Обзор представляет собой анализ материалов, посвященных изучению роли поражения сосудов головного мозга, нейродегенеративного процесса, хронической ишемии головного мозга в развитии болезни Альцгеймера и деменции. Генез мнестико-интеллектуальных расстройств обусловлен не столько первично-дегенеративными, сколько сосудистыми изменениями, особенно на уровне микроциркуляторного русла. Кроме того, нейроиммунный компонент усиливает цереброваскулярную патологию, что приводит к деменции и болезни Альцгеймера.
Ключевые слова: болезнь Альцгеймера, постинсультная деменция, когнитивные расстройство, мозговые антигены, бета-амилоид.
В последнее время проблема деменции выходит на одно из первых мест среди причин необратимой инвалидизации и смертности больных. Наряду с ростом сердечно-сосудистых заболеваний и инсультов, увеличивается и частота встречаемости сосудистой, в частности, постинсультной деменции.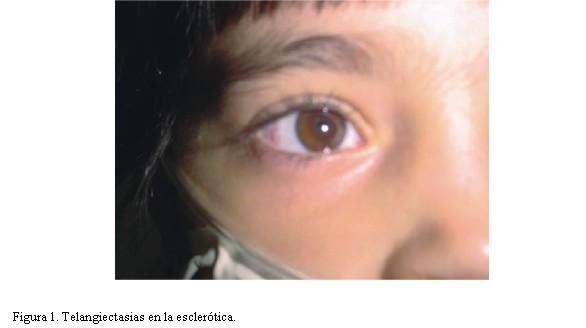 В наибольшей степени деменция затрагивает лиц пожилого и старческого возраста. В популяции людей 65-79 лет ее распространенность составляет 10-15%, а в возрасте 80 лет и старше достигает 20%. Почти у двух третей больных, выживших после инсульта, наблюдается выраженное в той или иной степени снижение когнитивной функции. Общая распространенность деменции у больных с инсультом составляет от 20% до 25%, при этом она увеличивается с возрастом и составляет около 14% у больных моложе 75 лет и приблизительно 32% — у больных старше 75 лет [1]. Вероятность развития деменции у больных с инсультом возрастает в 4-9 раз, по сравнению с лицами без инсульта. Повторные сосудистые эпизоды приводят к изменению иммунологического ответа на мозговые антигены головного мозга, что в свою очередь приводит к изменению гуморального ответа и формированию аутоиммунной реакции к антигенам мозга. Однако направленность иммунных реакций при острой ишемии головного мозга и хронической недостаточности мозгового кровообращения до настоящего времени недостаточно ясна [2].
В наибольшей степени деменция затрагивает лиц пожилого и старческого возраста. В популяции людей 65-79 лет ее распространенность составляет 10-15%, а в возрасте 80 лет и старше достигает 20%. Почти у двух третей больных, выживших после инсульта, наблюдается выраженное в той или иной степени снижение когнитивной функции. Общая распространенность деменции у больных с инсультом составляет от 20% до 25%, при этом она увеличивается с возрастом и составляет около 14% у больных моложе 75 лет и приблизительно 32% — у больных старше 75 лет [1]. Вероятность развития деменции у больных с инсультом возрастает в 4-9 раз, по сравнению с лицами без инсульта. Повторные сосудистые эпизоды приводят к изменению иммунологического ответа на мозговые антигены головного мозга, что в свою очередь приводит к изменению гуморального ответа и формированию аутоиммунной реакции к антигенам мозга. Однако направленность иммунных реакций при острой ишемии головного мозга и хронической недостаточности мозгового кровообращения до настоящего времени недостаточно ясна [2].
Болезнь Альцгеймера в последнее годы приобретает все большее медико-социальное значение. Это заболевание считается наиболее распространенной причиной деменции в пожилом и старческом возрасте. Затраты общества, связанные с болезнью Альцгеймера, сопоставимы с суммарными затратами на онкологические и кардиологические заболевания. Предполагается, что в настоящее время во всем мире болезнью Альцгеймера страдает от 17 до 25 млн. больных, а к 2050 году их число может увеличиться в 4 раза [3].
В настоящее время существует мнение, что болезнь Альцгеймера гетерогенна по своему происхождению: в одних случаях она носит наследственный характер, в других — возникает спорадический. Это заболевание может быть результатом сочетанного действия различных факторов, приводящих в конечном итоге сходным клиническим и
патоморфологическим изменениям [4]. R.Stewart
R.Stewart
рассматривая имеющиеся данные, свидетельствующие о связи артериальной гипотензии с болезнью Альцгеймера, подчеркивает, что характер данной связи требует уточнения. В частности, наличие деменции сопровождается снижением общего метаболизма, одним из проявлений которого и является пониженное артериальное давление [5].Повышение уровня гомоцистеина является одним из сосудистых факторов риска, на фоне гиперцистеинемии возрастает риск тромбозов, окклюзирующих поражений сосудов и инсульта. В последнее время было показано, что гиперцистеинемия отмечается как при сосудистой деменции, так и при болезни Альцгеймера, что приводит к активации механизмов воспаления, сопровождающихся нейротоксичностью, амилоидогенезом и
микроваскулярными расстройствами [6]. В 1994 году В.Хачински предложил использовать термин «сосудистые когнитивные расстройства» для обозначения нарушений высших мозговых функций вследствие цереброваскулярной патологии. Сосудистые когнитивные расстройства имеют характерные особенности патогенеза и клиники, а также течения, которые позволяют дифференцировать данный вид нарушений когнитивных функций от когнитивных нарушений нейродегенеративной природы, весьма распространенных в популяции [7].
Сосудистые когнитивные расстройства имеют характерные особенности патогенеза и клиники, а также течения, которые позволяют дифференцировать данный вид нарушений когнитивных функций от когнитивных нарушений нейродегенеративной природы, весьма распространенных в популяции [7].
По данным некоторых исследователей, значительные сосудистые изменения обнаруживаются у 48% пациентов болезнью Альцгеймера и у 33% лиц такого же возраста без ее признаков. По данным I.Skoog (2005), болезнь Альцгеймера обнаруживалась в 77% случаев сосудистой деменции и только в 17% случаев сосудистой деменции выявлялась изолированная сосудистая патология. Он указывает, что сочетание болезни Альцгеймера с цереброваскулярной патологией наблюдается у более, чем у 80% больных деменцией [8]. Связь деменции с сосудистым поражением головного мозга доказывается развитием когнитивных нарушений непосредственно после инсульта, либо после преходящих нарушений мозгового кровообращения. При различных нарушениях гомеостаза реакции иммунной системы обусловливают специфическую защиту организма. Однако в некоторых ситуациях иммунологические реакции способствуют развитию
При различных нарушениях гомеостаза реакции иммунной системы обусловливают специфическую защиту организма. Однако в некоторых ситуациях иммунологические реакции способствуют развитию
Лечение синдрома Луи Бар у детей в Израиле
ЛЕЧЕНИЕ В ИЗРАИЛЕ СНОВА ДОСТУПНО — ХАДАССА ЖДЕТ ПАЦИЕНТОВ
Клиника «Хадасса» продолжает оказывать помощь иностранным пациентам даже во время карантина. Сотрудники международного отдела помогают оперативно:
- согласовать разрешение в консульстве и Министерстве здравоохранения на прилет в Израиль;
- организовать очную консультацию с врачом и все необходимые процедуры;
- организовать госпитализацию в клинику.
ПО ВСЕМ ВОПРОСАМ ОБРАЩАЙТЕСЬ В НАШ ЕДИНЫЙ ЦЕНТР ЗАПИСИ ПО ТЕЛЕФОНУ 8(800)550-96-30
Здесь вы сможете пройти предварительную диагностику и получить удаленную консультацию ведущего израильского специалиста.
Синдром Луи-Бар, или атаксия телеангиэктазия — редкое иммунодефицитное заболевание наследственного характера. Впервые болезнь была описана в 1941 году, бельгийским врачом Дениз Луи-Бар. Точных статистических данных, говорящих о заболеваемости данной патологией нет. По некоторым данным, синдром Луи-Бар встречается у одного из 40 000 новорожденных. Мальчики и девочки болеют одинаково часто.
Впервые болезнь была описана в 1941 году, бельгийским врачом Дениз Луи-Бар. Точных статистических данных, говорящих о заболеваемости данной патологией нет. По некоторым данным, синдром Луи-Бар встречается у одного из 40 000 новорожденных. Мальчики и девочки болеют одинаково часто.
Синдром Луи-Бар характеризуется плохой координацией движений и расширенными кровеносными сосудами, иммунодефицитом и склонностью к развитию опухолей.
Учитывая тяжесть заболевания, необходимость симптоматического лечения и предупреждение онкологии, оптимальное решение при его диагностике — лечение в Израиле.
Предоставление высококвалифицированной медицинской помощи за умеренную плату — основная причина, по которой в Израиль обращаются множество иностранных граждан. Благодаря работе профессионалов люди навсегда забывают о своих недугах, даже самых тяжелых. Инновационное оборудование, применение новейших технологий и высокий уровень квалификации персонала — основные факторы успеха терапии в Израиле.
и начинается с точной диагностики. Это дает возможность врачу быть уверенным в правильности своих действий.
Все процедуры, которые проводятся с пациентом, соответствуют международным стандартам, а постоянные контроли качества не позволяют заниматься лечением, не имея достаточных знаний и умений.
Именно в Израиле успешно излечивают те болезни, которые в странах постсоветского пространства лечить еще не умеют или не могут из-за отсутствия надлежащего оборудования.
Кроме того, в Израиле берутся не просто за сложные болезни, но и за самые тяжелые случаи, а цены на лечение гораздо ниже, чем в аналогичных клиниках Европы и США.
Преимущества лечения в Хадассе
Детская медицина в Израиле является одним из приоритетных направлений, на которое выделяются огромные государственные средства. Это дает возможность оборудовать детский корпус Хадассы согласно с последними достижениями науки и техники.
К основным преимуществам лечения в Хадассе относятся:
- широкий спектр медицинских услуг — в состав детской клиники входят более тридцати подразделений;
- безболезненные программы диагностики — все диагностические алгоритмы учитывают детский страх перед медиками, а при инструментальной диагностике используются специальные мини-датчики, не причиняющие дискомфорта маленьким пациентам;
- высокотехнологичное оборудование и применение передовых лечебных технологий;
- уникальные курсы лечения — в клинику приглашают учителей, психологов, логопедов и других специалистов. Это делается с целью поддержки социальных навыков у детей;
- индивидуальный подход к каждому пациенту;
- разумная стоимость лечения.
 Это делается с целью поддержки социальных навыков у детей;
Это делается с целью поддержки социальных навыков у детей;Кроме того, детям всех возрастов предлагают прекрасно оборудованные палаты и русскоговорящего переводчика.
Для получения более точной информации о стоимости лечения и специальных предложениях нажмите кнопку УЗНАТЬ ТОЧНЫЕ ЦЕНЫСтоимость лечение синдрома Луи-Бар в Израиле
Несмотря на все преимущества лечения синдрома Луи-Бар, стоимость терапии в Израиле остается вполне доступной. Мы является официальными представителями клиники, поэтому вам не придется дополнительно оплачивать посреднические услуги. Вся оплата производится непосредственно в кассу учреждения. Узнать точную цену лечения можно позвонив по указанному на сайте номеру телефона или заполнив электронную анкету.
Проявления и симптомы синдрома Луи-Бар
К основным симптомам синдрома Луи-Бар относятся:
- атаксия;
- телеангиэктазия;
- инфекции дыхательных путей;
- опухолевые заболевания.

Атаксия — первое клиническое проявление заболевания, которое появляется в возрасте от пяти месяцев до трех лет. В большинстве случаев родители замечают неладное, когда малыш начинает ходить. Наблюдаются нарушения равновесия и походки, интенционный тремор, качания туловища и головы. Речь малыша становится невнятной, скандированной. Снижается мышечный тонус, исчезают сухожильные рефлексы, появляется нистагм и косоглазие.
Телеангинэктазии появляются в возрасте от трех до шести лет. Очень редко данное проявление возникает уже на первом месяце жизни малыша. Сосудистые звездочки представляют собой розовые или красноватые пятна, обусловленные расширением мелких сосудов кожи. Изначально образования появляются на конъюнктиве, затем на коже век, носа, лица и шеи, локтевых и коленных сгибов, предплечий, тыльной поверхности кистей и стоп. Также могут возникать на слизистой оболочке полости рта.
Основная причина инфекций дыхательных путей при синдроме Луи-Бар — поражение иммунной системы. У больных детей диагностируют хронические риниты, фарингиты, частые бронхиты и воспаление легких. Болезни отличаются длительным течением, стертостью границ между ремиссией и обострением, скудностью физикальных данных. Кроме того, острые эпизоды плохо поддаются антибактериальной терапии. Каждый подобный инфекционный эпизод несет смертельную опасность для больного атаксией-телеангиэктазией. Частые поражения легких ведут к развитию бронхоэктазов и пневмосклероза.
У больных детей диагностируют хронические риниты, фарингиты, частые бронхиты и воспаление легких. Болезни отличаются длительным течением, стертостью границ между ремиссией и обострением, скудностью физикальных данных. Кроме того, острые эпизоды плохо поддаются антибактериальной терапии. Каждый подобный инфекционный эпизод несет смертельную опасность для больного атаксией-телеангиэктазией. Частые поражения легких ведут к развитию бронхоэктазов и пневмосклероза.
Злокачественные опухолевые процессы среди больных синдромом Луи-Бар отмечаются в 1000 раз чаще, чем в общей популяции. Наиболее часто у пациентов диагностируют лимфомы и лейкемии. Больные атаксией-телеангиэктазией очень чувствительны к воздействию ионизирующего излучения, что полностью исключает применение лучевой терапии при лечении онкологических процессов.
Диагностика синдрома Луи-Бар
Диагностика синдрома Луи-Бар в Израиле проводится на основании данных клинической картины и дополнительных исследований. Как правило, из лабораторных и инструментальных методов применяют:
- общий анализ крови — снижается количество лейкоцитов;
- определение уровня альфа-фетопротеина — белок, концентрация которого повышается при синдроме Луи-Бар;
- иммунограмма — снижается количество иммуноглобулинов класса A, G, E;
- КТ и МРТ головного мозга, органов грудной клетки.

При необходимости больного синдромом Луи-Бар консультирует невролог, пульмонолог, ЛОР, иммунолог.
Лечение и прогноз синдрома Луи-Бар
Патогенетическим лечением синдрома Луи-Бар является пересадка вилочковой железы (тимуса). Операция выполняется в Израиле, в детском корпусе клиники Хадасса. Кроме того, пациенту показаны:
- своевременная антибиотикотерапия при развитии инфекционных осложнений;
- введение человеческого иммуноглобулина;
- курсы лечения препаратами, содержащими компоненты тимуса;
- неспецифическое лечение — правильное питание, умеренные физические нагрузки, сосудистая терапия.
Прогноз при синдроме Луи-Бар неблагоприятный. Средняя продолжительность жизни пациентов на сегодняшний день — 15 лет. Своевременное и полноценное лечение в Израиле увеличивает этот показатель. Наиболее распространенными причинами смерти больных являются инфекционные осложнения и злокачественные новообразования.
Синдром Луи-Бар — причины, симптомы, диагностика и лечение
Синдром Луи-Бар (атаксия-телеангиэктазия) — наследственное заболевание, проявляющееся мозжечковой атаксией, телеангиэктазиями кожи и конъюнктивы глаз, недостаточностью Т-клеточного звена иммунитета. Последнее приводит к тому, что синдром Луи-Бар сопровождается частыми респираторными инфекциями и склонностью к возникновению злокачественных опухолей. Диагностируется синдром Луи-Бар на основании анамнеза и клинической картины заболевания, данных иммунограммы, результатов офтальмологического и отоларингологического обследования, МРТ головного мозга и рентгенографии легких. В настоящее время синдром Луи-Бар не имеет специфического и эффективного лечения.
Последнее приводит к тому, что синдром Луи-Бар сопровождается частыми респираторными инфекциями и склонностью к возникновению злокачественных опухолей. Диагностируется синдром Луи-Бар на основании анамнеза и клинической картины заболевания, данных иммунограммы, результатов офтальмологического и отоларингологического обследования, МРТ головного мозга и рентгенографии легких. В настоящее время синдром Луи-Бар не имеет специфического и эффективного лечения.
Общие сведения
Синдром Луи-Бар впервые был описан в 1941 году во Франции. Нет точных данных о том, с какой частотой синдром Луи-Бар встречается среди современного населения. По некоторым сведениям эта цифра составляет 1 случай на 40 тысяч новорожденных. Однако, необходимо учитывать, что при смерти в раннем детском возрасте синдром Луи-Бар обычно остается не диагностированным. Известно, что заболевание одинаково часто поражает мальчиков и девочек. В неврологии синдром Луи-Бар относится к так называемым факомотозам — генетически обусловленным сочетанным поражениям кожи и нервной системы. В эту группу также входят нейрофиброматоз Реклингхаузена, ангиоматоз Стерджа—Вебера, туберозный склероз и др.
В эту группу также входят нейрофиброматоз Реклингхаузена, ангиоматоз Стерджа—Вебера, туберозный склероз и др.
Синдром Луи-Бар
Причины и патогенез
В основе патологических изменений, сопровождающих синдром Луи-Бар, лежат генетические нарушения, приводящие к развитию врожденной нейроэктодермальной дисплазии. Синдром Луи-Бар является аутосомно-рецессивным заболеванием, т. е. проявляется клинически только при получении рецессивного гена сразу от обоих родителей.
Морфологически атаксия-телеангиэктазия характеризуется дегенеративными изменениями тканей мозжечка, в частности потерей зернистых клеток и клеток Пуркинье. Дегенеративные изменения могут затрагивать зубчатое ядро мозжечка (nucleus dentatus), черную субстанцию (substantia nigra) и некоторые отделы коры головного мозга, иногда поражаются спиномозжечковые пути и задние столбы спинного мозга.
Синдром Луи-Бар сочетается с гипоплазией или аплазией тимуса, а также с врожденным дефицитом IgA и IgE. Эти нарушения в системе иммунитета приводят к появлению у пациентов частых инфекционных заболеваний, склонных к длительному и осложненному течению. Кроме того, иммунные нарушения могут потенцировать развитие злокачественных новообразований, зачастую берущих свое начало в структурах лимфоретикулярной системы.
Эти нарушения в системе иммунитета приводят к появлению у пациентов частых инфекционных заболеваний, склонных к длительному и осложненному течению. Кроме того, иммунные нарушения могут потенцировать развитие злокачественных новообразований, зачастую берущих свое начало в структурах лимфоретикулярной системы.
Клинические проявления синдрома Луи-Бар
Атаксия. Наиболее часто синдром Луи-Бар начинает проявляться клинически в возрасте от 5 месяцев до 3 лет. Во всех случаях заболевания синдром Луи-Бар манифестирует с появления мозжечковой атаксии, признаки которой становятся очевидными, когда ребенок начинает ходить. Наблюдаются нарушения равновесия и походки, дрожание во время двигательного акта (интенционный тремор), качание туловища и головы. Зачастую атаксия настолько выражена, что имеющий синдром Луи-Бар больной не может ходить. Мозжечковая атаксия сочетается с мозжечковой дизартрией, характеризующейся невнятной скандированной речью. Отмечается мышечная гипотония, снижение или полное исчезновение сухожильных рефлексов, нистагм, глазодвигательные нарушения и косоглазие.
Телеангиэктазии. В большинстве случаев появление сопровождающих синдром Луи-Бар телеангиэктазий происходит в возрасте от 3 до 6 лет. В некоторых случаях их возникновение отмечается в более поздний период и очень редко в течение первого месяца жизни. Телеангиэктазии (сосудистые звездочки) представляют собой имеющие различную форму красноватые или розовые пятнышки или разветвления. Они обусловлены расширением мелких сосудов кожи. Следует отметить, что телеангиэктазии могут быть проявлением многих других заболеваний (например, розацеа, СКВ, дерматомиозита, пигментной ксеродермы, хронического лучевого дерматита, мастоцитоза и пр.). Однако в сочетании с атаксией они дают специфическую для синдрома Луи-Бар клиническую картину.
Синдром Луи-Бар характеризуется изначальным возникновением телеангиэктазий на конъюнктиве глазного яблока, где они имеют вид «паучков». Затем сосудистые звездочки появляются на коже век, носа, лица и шеи, локтевых и коленных сгибов, предплечий, тыльной поверхности стоп и кистей. Телеангиэктазии могут также наблюдаться на слизистой оболочке мягкого и твердого неба. Наиболее выражены сосудистые звездочки в тех местах кожного покрова, где он подвергается воздействию солнечных лучей. В первую очередь это лицо, где телеангиэктазии образуют целые «пучки». При этом кожа теряет свою эластичность и становится плотной, что напоминает изменения, типичные для склеродермии.
Телеангиэктазии могут также наблюдаться на слизистой оболочке мягкого и твердого неба. Наиболее выражены сосудистые звездочки в тех местах кожного покрова, где он подвергается воздействию солнечных лучей. В первую очередь это лицо, где телеангиэктазии образуют целые «пучки». При этом кожа теряет свою эластичность и становится плотной, что напоминает изменения, типичные для склеродермии.
Кожные проявления атаксии-телеангиэктазии могут включать появление веснушек и пятен цвета кофе с молоком, участков обесцвеченной кожи. Наличие гипо- и гиперпигментаций делает кожные симптомы синдрома Луи-Бар схожими с клиникой пойкилодермии. У многих больных отмечается сухость кожи и участки гиперкератоза. Может наблюдаться гипертрихоз, ранняя седина волос, кожные элементы, напоминающие акне или проявления псориаза.
Инфекции дыхательных путей. Характеризующее синдром Луи-Бар поражение иммунной системы приводит к возникновению частых рецидивирующих инфекций дыхательных путей и уха: хронических ринитов, фарингитов, бронхитов, пневмоний, отитов, синуситов. Их особенностями являются: стертость границ между периодом обострения и ремиссии, скудность физикальных данных, плохая чувствительность к антибактериальной терапии и длительное течение. Каждая подобная инфекция может стать для больного атаксией-телеангиэктазией смертельно опасной. Частые заболевания легких приводят к развитию бронхоэктазов и пневмосклероза.
Их особенностями являются: стертость границ между периодом обострения и ремиссии, скудность физикальных данных, плохая чувствительность к антибактериальной терапии и длительное течение. Каждая подобная инфекция может стать для больного атаксией-телеангиэктазией смертельно опасной. Частые заболевания легких приводят к развитию бронхоэктазов и пневмосклероза.
Злокачественные новообразования. Среди пациентов, имеющих синдром Луи-Бар, злокачественные опухолевые процессы отмечаются в 1000 раз чаще, чем в среднем у населения. Наиболее распространенными среди них являются лейкемия и лимфома. Особенностью онкопатологии в случае синдрома Луи-Бар является повышенная чувствительность пациентов к воздействию ионизирующего излучения, что полностью исключает применение лучевой терапии при их лечении.
Диагностика синдрома Луи-Бар
Постановка диагноза атаксии-телеангиэктазии требует комплексного подхода, учитывающего анамнез заболевания, его клинические проявления, данные иммунологических и инструментальных исследований, а также результаты ДНК-диагностики.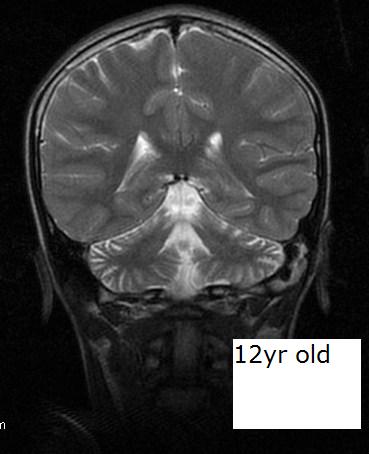 Пациент с подозрением на синдром Луи-Бар должен пройти обследование не только у невролога, но и у дерматолога, отоларинголога, офтальмолога, иммунолога, пульмонолога, онколога.
Пациент с подозрением на синдром Луи-Бар должен пройти обследование не только у невролога, но и у дерматолога, отоларинголога, офтальмолога, иммунолога, пульмонолога, онколога.
Лабораторная диагностика синдрома Луи-Бар включает клинический анализ крови, в котором у 1/3 пациентов наблюдается снижение количества лимфоцитов. Обязательно проводится исследование уровня иммуноглобулинов крови, которое выявляет значительное снижение IgA и IgE, в 10-12% случаев IgG. Примерно у 40% пациентов синдром Луи-Бар сопровождается аутоиммунными реакциями, о которых свидетельствует наличие аутоантител к митохондриям, тиреоглобулину, иммуноглобулинам.
Из инструментальных способов диагностики синдрома Луи-Бар могут применяться: УЗИ тимуса, МРТ головного мозга, фарингоскопия, риноскопия, рентгенография легких. При помощи УЗИ диагностируется аплазия или гипоплазия тимуса. МРТ головного мозга выявляет атрофию мозжечка, расширение IV желудочка. Рентгенография легких необходима для диагностики очаговой или крупозной пневмонии, выявления очагов пневмосклероза и бронхоэктатических изменений.
Синдром Луи-Бар следует дифференцировать с атаксией Фридрейха, болезнью Рандю-Ослера, атаксией Пьера-Мари, болезнью Гиппеля-Линдау и др.
Лечение и прогноз синдрома Луи-Бар
К сожалению, эффективные методы лечения синдрома Луи-Бар до настоящего времени остаются предметом поиска. В современной медицине возможно применение лишь паллиативного симптоматического лечения соматических и иммунологических нарушений. Продлению жизни пациентов, имеющих синдром Луи-Бар, способствует иммунокоррегирующая терапия препаратами тимуса и гамма-глобулином, витаминотерапия в высоких дозировках и интенсивная терапия любого инфекционного процесса. По показаниям применяют противовирусные препараты, антибиотики широкого спектра действия, противогрибковые средства, глюкокортикостероиды.
В связи с отсутствием эффективных способов лечения синдром Луи-Бар имеет неблагоприятный прогноз как для выздоровления, так и для жизни. Больные этим заболеванием редко доживают до 20 лет. В большинстве случаев они погибают от инфекционных осложнений и онкологических заболеваний.
В большинстве случаев они погибают от инфекционных осложнений и онкологических заболеваний.
ФАРМАТЕКА » Необычные кожные проявления при атаксии–телеангиоэктазии (синдром Луи-Бар)
В статье приводится описание необычных кожных проявлений у девочки 5 лет с врожденной атаксией-телеангиоэктазией (синдром Луи-Бар), имеющей признаки липоидного некробиоза. Синдром Луи-Бар – редкая генетически обусловленная патология, выражающаяся в сочетанном поражении кожи и нервной системы. Диагностика синдрома обычно крайне затруднена ввиду редкой встречаемости и ограниченной информации у врачей смежных специальностей. В статье описан порядок проведения диагностических мероприятий и дифференциальной диагностики с целью установления диагноза. Приведены данные гистологического исследования. Отмечено, что данный случай представляет особый интерес, т.к. в доступной литературе не было найдено описаний подобных кожных проявлений при этом редком генетическом заболевании.
Синдром Луи-Бар (D. Louis-Bar) впервые был описан в 1941 г. во Франции. Частота встречаемости составляет 1 случай на 40 тыс. новорожденных. Известно, что заболевание одинаково часто поражает мальчиков и девочек. В неврологии синдром Луи-Бар относится к т.н. факоматозам — генетически обусловленным сочетанным поражениям кожи и нервной системы. В эту группу также входят нейрофиброматоз Реклингхаузена, ангиоматоз Стерджа–Вебера, туберозный склероз и др. В основе патологических изменений, сопровождающих синдром Луи-Бар, лежат генетические нарушения, приводящие к развитию врожденной нейроэктодермальной дисплазии. Синдром Луи-Бар является аутосомно-рецессивным заболеванием, т.е. проявляется клинически только при получении рецессивного гена сразу от обоих родителей [1]. Наиболее часто синдром Луи-Бар начинает проявляться в возрасте от 5 месяцев до 3 лет. При этом отмечается появление мозжечковой атаксии, признаки которой становятся очевидными, когда ребенок начинает ходить. Наблюдаются нарушения равновесия и походки, дрожание, качание туловища и головы во время движения.
Louis-Bar) впервые был описан в 1941 г. во Франции. Частота встречаемости составляет 1 случай на 40 тыс. новорожденных. Известно, что заболевание одинаково часто поражает мальчиков и девочек. В неврологии синдром Луи-Бар относится к т.н. факоматозам — генетически обусловленным сочетанным поражениям кожи и нервной системы. В эту группу также входят нейрофиброматоз Реклингхаузена, ангиоматоз Стерджа–Вебера, туберозный склероз и др. В основе патологических изменений, сопровождающих синдром Луи-Бар, лежат генетические нарушения, приводящие к развитию врожденной нейроэктодермальной дисплазии. Синдром Луи-Бар является аутосомно-рецессивным заболеванием, т.е. проявляется клинически только при получении рецессивного гена сразу от обоих родителей [1]. Наиболее часто синдром Луи-Бар начинает проявляться в возрасте от 5 месяцев до 3 лет. При этом отмечается появление мозжечковой атаксии, признаки которой становятся очевидными, когда ребенок начинает ходить. Наблюдаются нарушения равновесия и походки, дрожание, качание туловища и головы во время движения.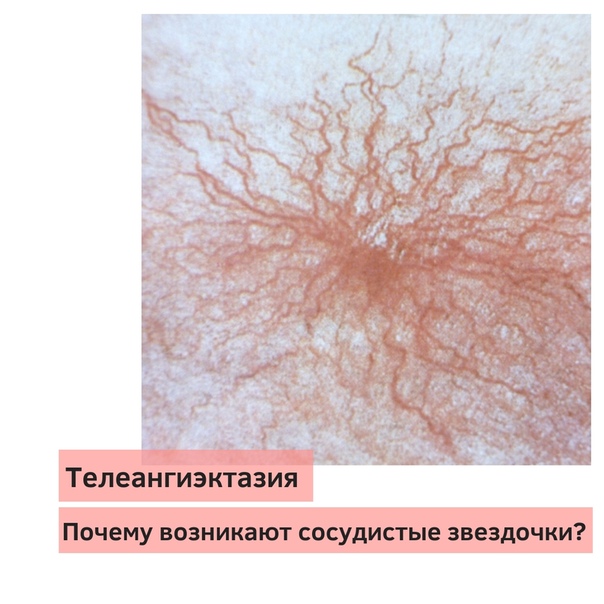 Характерна мозжечковая дизартрия, характеризующаяся невнятной скандированной речью. Отмечаются мышечная гипотония, снижение или полное исчезновение сухожильных рефлексов, нистагм, глазодвигательные нарушения и косоглазие. Имеется склонность к инфекционным заболеваниям: частые острые респираторно-вирусные инфекции, пневмонии, имеющие затяжное течение и плохо поддающиеся лечению. Нередки опухоли яичников, желудка, кожи.
Характерна мозжечковая дизартрия, характеризующаяся невнятной скандированной речью. Отмечаются мышечная гипотония, снижение или полное исчезновение сухожильных рефлексов, нистагм, глазодвигательные нарушения и косоглазие. Имеется склонность к инфекционным заболеваниям: частые острые респираторно-вирусные инфекции, пневмонии, имеющие затяжное течение и плохо поддающиеся лечению. Нередки опухоли яичников, желудка, кожи.
Телеангиэктазии появляются в возрасте от 3 до 6 лет и первоначально возникают на конъюнктиве глазного яблока, затем на коже век, носа, лица и шеи, локтевых и коленных сгибах, предплечьях, тыльной поверхности стоп и кистей. Телеангиэктазии могут также наблюдаться на слизистой оболочке мягкого и твердого неба, но наиболее выражены в тех местах кожного покрова, которые подвергаются воздействию солнечных лучей. В первую очередь это лицо, где телеангиэктазии образуют целые «пучки». Кожа при этом теряет свою эластичность и становится плотной, что напоминает изменения, типичные для склеродермии. Из других кожных проявлений встречаются веснушки, пятна цвета «кофе с молоком», участки обесцвеченной кожи. Наличие гипо- и гиперпигментаций делает кожные симптомы синдрома Луи-Бар схожими с клиникой пойкилодермии. У многих больных отмечается сухость кожи и участки гиперкератоза. Могут наблюдаться гипертрихоз, ранняя седина волос, кожные элементы, напоминающие акне, или проявления псориаза [2].
Из других кожных проявлений встречаются веснушки, пятна цвета «кофе с молоком», участки обесцвеченной кожи. Наличие гипо- и гиперпигментаций делает кожные симптомы синдрома Луи-Бар схожими с клиникой пойкилодермии. У многих больных отмечается сухость кожи и участки гиперкератоза. Могут наблюдаться гипертрихоз, ранняя седина волос, кожные элементы, напоминающие акне, или проявления псориаза [2].
Лабораторная диагностика синдрома Луи-Бар включает клинический анализ крови, в котором у трети пациентов наблюдается снижение количества лимфоцитов. Обязательно проводится исследование уровня иммуноглобулинов (Ig) крови, которое выявляет значительное снижение IgA и IgЕ, в 10–12% случаев IgG. Примерно у 40% пациентов синдром Луи-Бар сопровождается аутоиммунными реакциями, о которых свидетельствует наличие аутоантител к митохондриям, тиреоглобулину, иммуноглобулинам.
Из инструментальных способов диагностики синдрома Луи-Бар могут применяться ультразвуковое исследование тимуса, при котором может быть диагностирована аплазия или гипоплазия тимуса; магнитно-резонансная томография головного мозга с целью выявления атрофии мозжечка, расширения IV желудочка; фарингоскопия, риноскопия. Также выполняется рентгенография легких для диагностики очаговой или крупозной пневмонии, выявления очагов пневмосклероза и бронхоэктатических изменений [3, 4]. Синдром Луи-Бар следует дифференцировать с атаксией Фридрейха, болезнью Рандю–Ослера, атаксией Пьера–Мари, синдромом Гиппеля–Линдау и др.
Также выполняется рентгенография легких для диагностики очаговой или крупозной пневмонии, выявления очагов пневмосклероза и бронхоэктатических изменений [3, 4]. Синдром Луи-Бар следует дифференцировать с атаксией Фридрейха, болезнью Рандю–Ослера, атаксией Пьера–Мари, синдромом Гиппеля–Линдау и др.
Выявление синдрома Луи-Бар требует комплексного подхода, учитывающего анамнез заболевания, его клинические проявления, данные иммунологических и инструментальных исследований, а также результаты ДНК-диагностики. Пациент с подозрением на синдром Луи–Бар должен пройти обследование не только у невролога, но и у дерматолога, отоларинголога, офтальмолога, иммунолога, пульмонолога, онколога.
К сожалению, эффективные методы лечения синдрома Луи-Бар до настоящего времени остаются предметом поиска. В современной медицине возможно применение лишь паллиативного симптоматического лечения соматических и иммунологических нарушений. Продлению жизни пациентов, имеющих синдром Луи-Бар, способствует иммунокорригирующая терапия препаратами тимуса (Т-активин) и гамма-глобулином, витаминотерапия в высоких дозировках и интенсивная терапия любого инфекционного процесса.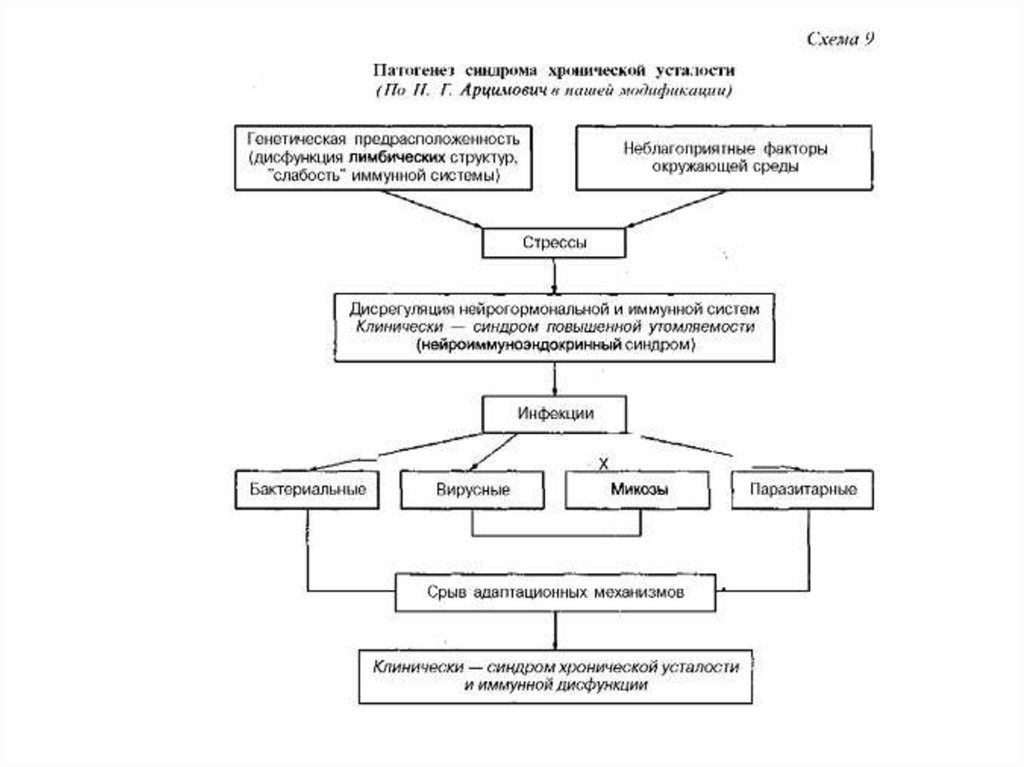 По показаниям применяют противовирусные препараты, антибиотики широкого спектра действия, противогрибковые средства, глюкокортикостероиды.
По показаниям применяют противовирусные препараты, антибиотики широкого спектра действия, противогрибковые средства, глюкокортикостероиды.
Под нашим наблюдением находилась девочка М. 5 лет, у которой в год и 3 месяца обнаружилась атаксия. Приблизительно в двухлетнем возрасте появились расширенный сосуд на правой конъюнктиве, папулезная сыпь на нижних конечностях. В 2012 г. на основании клинической картины, наличия первичного иммунодефицита (снижение уровней IgA, -G), ДНК-диагностики (наличие мутированного гена АТМ) был поставлен диагноз «атаксия–телеангиэктазия». При гистологическом исследовании биоптатов кожи был обнаружен продуктивный гранулематозный дерматит, вероятно инфекционной природы, который не был подтвержден ни клинически, ни лабораторно. Девочка получала заместительную терапию препаратами иммуноглобулинов, биологическими агентами, симптоматическими средствами, местно кремом, содержащим такролимус. В 2014 г. проведено повторное гистологическое исследование кожных высыпаний, по результатам которого было получено заключение: саркоидозный дерматит. Проведена рентгеновская компьютерная томография органов брюшной и грудной полостей, которая не выявила патологии. С 2015 г. отмечено ухудшение кожного процесса с увеличением размера высыпаний и появления в их центре изъязвлений без болезненных ощущений и лихорадки. Проведено очередное гистологическое исследование на кафедре анатомии МГМУ. И.М. Сеченова, которое выявило множественные сливающиеся эпителиоидноклеточные и гигантоклеточные гранулемы со скоплением макрофагов, отдельных гигантских многоядерных клеток типа Пирогова–Лангханса, а также лимфоцитов. Гранулемы локализуются в дерме и окружены валом воспалительного лимфогистиоцитарного инфильтрата. Часть гранулем имеют в центре казеозный некроз. Было получено заключение о необходимости проведения дифференциального диагноза между саркоидозом, микобактериозом и туберкулезом. При осмотре: состояние тяжелое по основному заболеванию. Жалобы на шаткость походки, кожные высыпания. Местный статус: на нижних конечностях, ягодицах имеются бляшки округлых овальных очертаний диаметром 5–8 см.
Проведена рентгеновская компьютерная томография органов брюшной и грудной полостей, которая не выявила патологии. С 2015 г. отмечено ухудшение кожного процесса с увеличением размера высыпаний и появления в их центре изъязвлений без болезненных ощущений и лихорадки. Проведено очередное гистологическое исследование на кафедре анатомии МГМУ. И.М. Сеченова, которое выявило множественные сливающиеся эпителиоидноклеточные и гигантоклеточные гранулемы со скоплением макрофагов, отдельных гигантских многоядерных клеток типа Пирогова–Лангханса, а также лимфоцитов. Гранулемы локализуются в дерме и окружены валом воспалительного лимфогистиоцитарного инфильтрата. Часть гранулем имеют в центре казеозный некроз. Было получено заключение о необходимости проведения дифференциального диагноза между саркоидозом, микобактериозом и туберкулезом. При осмотре: состояние тяжелое по основному заболеванию. Жалобы на шаткость походки, кожные высыпания. Местный статус: на нижних конечностях, ягодицах имеются бляшки округлых овальных очертаний диаметром 5–8 см. Периферическая часть бляшек красновато-сиреневатого цвета, центральная часть западает желтовато-буроватого цвета. На поверхности легкое шелушение, желтые корки и многочисленные телеангиоэктазии (рис. 1, 2).
Периферическая часть бляшек красновато-сиреневатого цвета, центральная часть западает желтовато-буроватого цвета. На поверхности легкое шелушение, желтые корки и многочисленные телеангиоэктазии (рис. 1, 2).
С учетом клинических проявлений кожного процесса, гистологических исследований, отсутствия данных за саркоидоз и туберкулез можно думать о липоидном некробиозе. Причем отсутствие липоидных отложений в биоптате говорит в пользу гранулематоза Мишера–Ледера, который, впрочем, также, по мнению ряда гистологов, представляет собой липоидный некробиоз [5]. Данный случай представляет особый интерес, т.к. в доступной литературе мы не нашли описания подобных кожных проявлений при этом редком генетическом заболевании. Тем более что врачи других специальностей не знакомы с редкой кожной патологией, что вызвало неправильную трактовку гистологического исследования. Фибриноидная дегенерация коллагена, имеющая место при липоидном некробиозе, скорее всего была принята за казеозный некроз, что и привело к заключению о микробной этиологии высыпаний, хотя тщательное обследование больной на туберкулез дало отрицательный результат.
И.Ю. Голоусенко – д.м.н., доцент, ГБОУ ВПО МГМСУ им А.И. Евдокимова Минздрава России, Москва; e-mail: [email protected]
Атаксия и телеангиэктазия (синдром Луи-Бар) — Консультант по дерматологии
Уверены ли вы в диагнозе?
На что следует обратить внимание в истории
Пациенты обычно обращаются с неврологическими нарушениями; т. е. атаксия с первых месяцев жизни. Атаксия туловища (колебания туловища) может проявляться на 5-6 месяцах жизни, в то время как атаксия походки появляется, когда пораженный ребенок начинает ходить, с частыми падениями. Глазодвигательная апраксия (медленные или отсутствующие произвольные движения глаз) проявляется со второго года жизни.Нейродегенерация прогрессирует, что приводит к инвалидной коляске во втором десятилетии жизни (рис. 1).
Глазодвигательная апраксия (медленные или отсутствующие произвольные движения глаз) проявляется со второго года жизни.Нейродегенерация прогрессирует, что приводит к инвалидной коляске во втором десятилетии жизни (рис. 1).
Рисунок 1.
Представление первого признака этой сущности (атаксия), которая может возникнуть даже в течение первого года жизни.
Многие пациенты (около 70%) страдают рецидивирующими синопульмональными инфекциями, в результате чего большинство из них умирают в подростковом возрасте. Вторая причина смерти — возникновение Т-клеточного лейкоза или лимфомы или, реже, солидных опухолей.Резкая радиогиперчувствительность, связанная с этим заболеванием, затрудняет противоопухолевое лечение, поэтому обычно используются ослабленные протоколы.
Характерные результаты медицинского осмотра
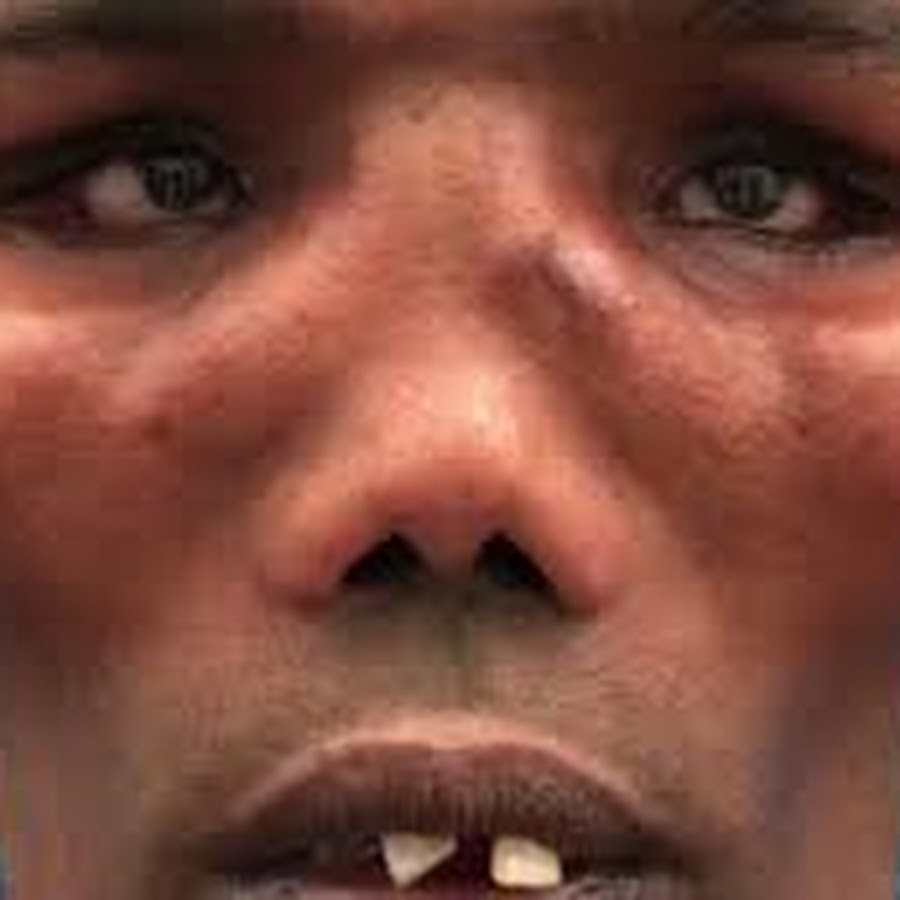
Вторым признаком заболевания является наличие глазно-кожных телеангиэктазий (рис. 2), которые присутствуют более чем у 90% пациентов.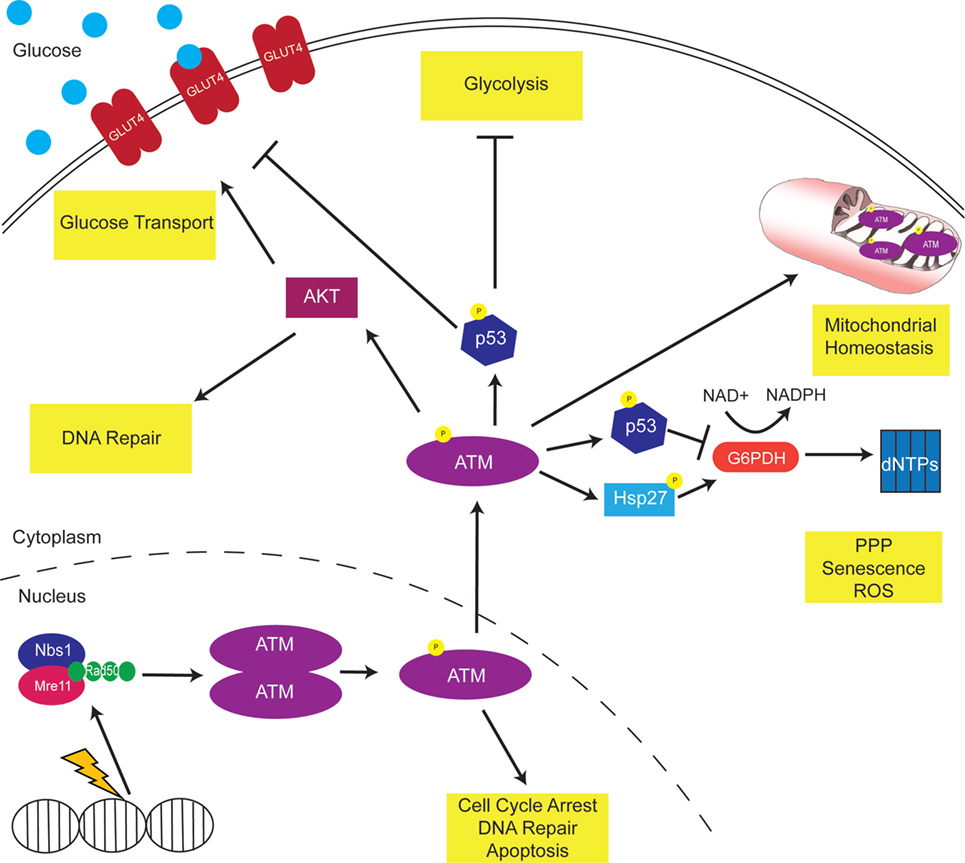 На коже часто наблюдаются пятна с молоком, гранулемы, гипо- и гиперпигментированные участки, а также раннее поседение волос. Прогероидный аспект характерен для многих пациентов.
На коже часто наблюдаются пятна с молоком, гранулемы, гипо- и гиперпигментированные участки, а также раннее поседение волос. Прогероидный аспект характерен для многих пациентов.
Ожидаемые результаты диагностических исследований
Диагноз подтверждается высоким уровнем сывороточного альфафетопротеина и повышенной частотой хромосомных разрывов, как спонтанных, так и вызванных гамма-излучением и радиомиметическими препаратами, а также отсутствием белка ATM (как показывает анализ вестерн-блоттинга). Ядерно-магнитно-резонансная томография (ЯМР) демонстрирует неспецифическую атрофию мозжечка с вовлечением червя и / или полушарий, а также увеличение четвертого желудочка.
Подтверждение диагноза
Подтверждение диагноза у пациентов с более мягким фенотипом иногда затруднено; Дифференциальный диагноз по сравнению с AT-LD (атаксия, похожая на телеангиэктазию), AOA1 и AOA2 (атаксия с глазодвигательной апраксией 1 и 2 типа (AOA1, AOA2) в основном проводится в лаборатории на основе нескольких Вестерн-блоттингов, выполненных со специфическим Антитела. Молекулярный анализ гена ATM, выполняемый денатурирующей высокоэффективной жидкостной хроматографией (DHPLC), прямым секвенированием и / или мультиплексным лигированием-зависимым анализом зонда (MLPA), дает окончательное подтверждение диагноза.
Молекулярный анализ гена ATM, выполняемый денатурирующей высокоэффективной жидкостной хроматографией (DHPLC), прямым секвенированием и / или мультиплексным лигированием-зависимым анализом зонда (MLPA), дает окончательное подтверждение диагноза.
Кто подвержен риску развития этого заболевания?
Атаксия Телеангиэктазия наследуется по аутосомно-рецессивному типу, что означает, что два здоровых родителя, оба носители одной мутации ATM, при каждой беременности имеют 25% -ный риск рождения больного ребенка. Как и в случае других аутосомно-рецессивных заболеваний, кровное родство является фактором риска. Атаксия Телеангиэктазия присутствует во всем мире.
Что является причиной болезни?
Этиология
Атаксия телеангиэктазия вызвана двуаллельными мутациями в гене ATM
Патофизиология
Ген ATM контролирует клеточный цикл и клеточную реакцию на окислительный стресс и на двухцепочечные разрывы ДНК, наиболее токсичные повреждения ДНК. Отсутствие белка ATM приводит к нарушению регуляции клеточного цикла, отсутствию репарации повреждений ДНК и увеличению скорости хромосомных повреждений.
Отсутствие белка ATM приводит к нарушению регуляции клеточного цикла, отсутствию репарации повреждений ДНК и увеличению скорости хромосомных повреждений.
Системные последствия и осложнения
Атаксия Телеангиэктазия — мультисистемное заболевание с неврологическими, иммунологическими и эндокринными особенностями. Наиболее ярким осложнением является рецидив синопульмональных инфекций из-за иммунодефицита и / или затрудненного глотания, вызывающего аспирацию (ab ingestis pneumoni). Периодическое введение иммуноглобулинов и постоянная физиотерапевтическая поддержка могут отсрочить последствия этих дефектов.
Варианты лечения
Профилактика первичных проявлений болезни пока безуспешна. В последние годы были предприняты попытки симптоматической терапии с минимальным или нулевым эффектом. В последнее время было показано, что немногие пациенты чувствительны к терапии кортикостероидами. В настоящее время проводятся терапевтические испытания бета- и декса-метазона с целью увеличения числа пролеченных пациентов и поиска оптимальной дозировки, которая могла бы уменьшить побочные эффекты длительного приема кортикостероидов.
Оптимальный подход к лечению этого заболевания
Пациенты должны постоянно проходить физиотерапию, особенно для облегчения респираторных заболеваний. Также полезна гипотерапия, особенно для улучшения атаксической позы. Инвалидные коляски необходимы к 10-12 годам у пациентов с классической, тяжелой формой заболевания.
Управление пациентами
Наблюдение за пациентами обязательно как с неврологическими, так и с иммунологическими проблемами. Тем не менее, из-за отсутствия эффективной терапии прогрессирующую кахексию остановить невозможно.Если присутствует злокачественная опухоль, необходимо использовать ослабленные терапевтические протоколы.
Необычные клинические сценарии, которые следует учитывать при ведении пациентов
В последние годы число пациентов, диагностированных во взрослом возрасте, увеличилось за счет совершенствования диагностических инструментов. У этих пациентов наблюдается позднее начало (по крайней мере, после полового созревания) неврологических особенностей, обычно начинающихся с периферических поражений (гипотония, тремор, хорея).
У этих пациентов наблюдается позднее начало (по крайней мере, после полового созревания) неврологических особенностей, обычно начинающихся с периферических поражений (гипотония, тремор, хорея).
Атаксия становится очевидной через несколько лет после начала и может оставаться легкой в течение многих лет, обеспечивая вполне нормальную жизнь, а также размножение.Учитывая мягкий фенотип, дозировка сывороточного альфафетопротеина, по нашему мнению, является обязательной для всех взрослых пациентов с идиопатической атаксией и отрицательными молекулярными результатами по отношению к другим формам атаксии взрослых.
У классических пациентов улучшение качества жизни за последние два десятилетия привело к увеличению продолжительности жизни; как следствие, число пациентов с АТ, страдающих опухолями, увеличивается, и часто описываются солидные опухоли (менингиома, карциномы груди и желудка и т. д.).Онкологи должны быть осторожны при лечении этих пациентов, используя упрощенные протоколы, а также сокращая количество радиологических анализов.
Какие есть доказательства?
Broccoletti, T., Del Giudice, E, Cirillo, E, Giardino, G, Ginocchio, VM, Bruscoli, S. «Эффективность очень низких доз бетаметазона на неврологические симптомы при атаксии-телеангиэктазии». Eur J Neurol. 2010 sep 14. (Целью этого исследования было оценить минимальную терапевтически эффективную дозировку бетаметазона при неврологических симптомах AT у шести чувствительных пациентов с AT.Авторы обнаружили, что бетаметазон эффективен при А-Т в минимальной дозировке, и предоставили доказательства класса IIIA, что бетаметазон в очень низких дозах эффективен для улучшения неврологических симптомов у пациентов, страдающих атаксией телеангиэктазией.)
Буони, С., Заннолли, Р., Соррентино, Л., Фойс, А. «Бетаметазон и улучшение неврологических симптомов у пациентов с атаксией-телеангиэктазией». Arch Neurol. об. 63. 2006. С. 1479–82. (Это первый отчет о контроле симптомов центральной нервной системы у пациентов с атаксией-телеангиэктазией. В нем описывается эффективность кортикостероидной терапии в отношении симптомов центральной нервной системы у ребенка с атаксией-телеангиэктазией, у которого неврологические симптомы улучшились, когда ему иногда давали бетаметазон для лечения приступов астматического бронхита.)
В нем описывается эффективность кортикостероидной терапии в отношении симптомов центральной нервной системы у ребенка с атаксией-телеангиэктазией, у которого неврологические симптомы улучшились, когда ему иногда давали бетаметазон для лечения приступов астматического бронхита.)
Чун, ХХ, Гатти, РА. «Атаксия-телеангиэктазия, развивающийся фенотип». Ремонт ДНК. об. 3. 2004. С. 1187–96. (В этой обзорной статье описаны элементы атаксии-телеангиэктазии, обобщены лабораторные данные и определены характеристики отличных от других аутосомно-рецессивных мозжечковых атаксий (ARCA), таких как атаксия Фридрейха, дефицит Mre11 (AT-подобная болезнь) и глазодвигательная апраксия 1 ( дефицит апратаксина) и 2 (дефицит сенатаксина) при молекулярном тестировании.)
Дролет, Б.А., Дроле, Б, Звулунов, А, Якобсен, Р, Трой, Дж., Эстерли, Н.Б. «Кожные гранулемы как признак атаксии-телеангиэктазии». Дерматология. об. 194. 1997. pp. 273-5. (Телеангиэктазия является классическим кожным признаком атаксии-телеангиэктазии (AT) и часто является физическим признаком, указывающим на диагноз. Авторы сообщают о пациенте, у которого неинфекционные кожные гранулемы были характерным кожным признаком AT, и обсуждают синдромы иммунодефицита, связанные с похожие кожные гранулемы.)
Авторы сообщают о пациенте, у которого неинфекционные кожные гранулемы были характерным кожным признаком AT, и обсуждают синдромы иммунодефицита, связанные с похожие кожные гранулемы.)
Лавин М.Ф., Шилох Ю. «Генетический дефект при атаксии-телеангиэктазии». Ann Rev Immunol. об. 15. 1997. С. 177-202. Эта статья представляет собой подробный обзор генетического дефекта при атаксии и телеангиэктазии и его роли в хромосомной нестабильности и радиочувствительности, столь характерных для этого заболевания, с акцентом на понимание природы дефекта, полученное в результате недавней идентификации с помощью позиционного клонирования ответственных за это заболевание. ген, банкомат.)
Леуцци, В, Элли, Р, Антонелли, А, Чесса, Л, Кардона, Ф, Маркучи, Л, Петринелли, П.«Неврологические и цитогенетические исследования у пациентов с ранней атаксией-телеангиэктазией». Eur J Pediatr. об. 152. 1993. pp. 609-12. (Клинический диагноз АТ затруднен в возрасте до 4 лет. Авторы сообщают клинические и цитогенетические данные по трем пациентам с ранним началом и ранним диагнозом АТ в возрасте 12, 18 и 22 месяцев, соответственно. )
)
Льюис, РФ, Ледерман, HM, Кроуфорд, TO. «Глазные моторные аномалии при атаксии и телеангиэктазии». Ann Neurol. об.46. 1999. С. 287–95. (Хотя аномальные движения глаз являются характерным признаком атаксической телеангиэктазии, характеристики глазодвигательной дисфункции описаны только в небольших группах пациентов. Авторы исследовали движения глаз у 56 пациентов с атаксической телеангиэктазией, получив электроокулографические записи движений глаз у 33 пациентов. .)
Луи-Бар, Д. «С прогрессирующим синдромом, сопровождающимся телангиэктазиями, кожными капиллярами и конъюнктивами, симметричными наивными и неприятными проблемами». Confin Neurol (Базель). об. 4. 1941. С. 432–42. (Первое описание пациентов с АТ было опубликовано Силлабой и Хеннером в 1926 году. Они сообщили о трех чешских братьях-подростках с прогрессирующим хореоатетозом и поразительной глазной телеангиэктазии, как имеющих вариант семейного двойного атетоза Рамси Ханта. В 1941 году было опубликовано второе клиническое описание. Луи-Бар, в отношении 9-летнего бельгийского мальчика с прогрессирующей мозжечковой атаксией и обширной кожной телеангиэктазией, распределенной в виде невоидных пятен; не сообщалось ни о семейном анамнезе, ни о патологических исследованиях.Автор идентифицировал синдром как ранее не описанную сущность, принадлежащую к факоматозам, либо вариант синдрома Стерджа-Вебера, либо отдельную новую сущность.)
Луи-Бар, в отношении 9-летнего бельгийского мальчика с прогрессирующей мозжечковой атаксией и обширной кожной телеангиэктазией, распределенной в виде невоидных пятен; не сообщалось ни о семейном анамнезе, ни о патологических исследованиях.Автор идентифицировал синдром как ранее не описанную сущность, принадлежащую к факоматозам, либо вариант синдрома Стерджа-Вебера, либо отдельную новую сущность.)
Савицкий, К., Бар-Шира, А., Гилад, С., Ротман, Г., Зив, Й, Ванагайте, Л. «Один ген Ataxia Telangiectasia с продуктом, аналогичным киназе PI-3». Наука. об. 268. 1995. С. 1749-53. (Ген ATM, который мутирован при аутосомно-рецессивном заболевании ataxia telangiectasia [AT], был идентифицирован путем позиционного клонирования на хромосоме 11q22-23.Открытие ATM улучшило понимание AT и родственных синдромов и может позволить идентифицировать гетерозиготы AT, которые подвержены повышенному риску рака.)
Copyright © 2017, 2013 ООО «Поддержка принятия решений в медицине». Все права защищены.
Ни один спонсор или рекламодатель не участвовал, не одобрял и не платил за контент, предоставляемый Decision Support in Medicine LLC. Лицензионный контент является собственностью DSM и защищен авторским правом.
Атаксия и телеангиэктазия — NORD (Национальная организация редких заболеваний)
УЧЕБНИКИ
Gatti RA.Атаксия-телеангиэктазия. В: Справочник НОРД по редким заболеваниям. Липпинкотт Уильямс и Уилкинс. Филадельфия, Пенсильвания. 2003: 606-6.
Джонс KL. Распознаваемые модели пороков развития человека Смита. 5-е изд. Филадельфия, Пенсильвания; W.B. Компания Сондерс; 1997: 196-197.
Behrman RE, et al., Eds. Учебник педиатрии Нельсона. 15-е изд. Филадельфия, Пенсильвания; W.B. Компания Сондерс; 1996: 576, 1709.
Buyse ML. Энциклопедия врожденных пороков. Довер, Массачусетс; Blackwell Scientific Publications, Inc .; 1990: 205-207.
Горлин Р.Дж. и др., Ред. Синдромы головы и шеи. 3-е изд. Нью-Йорк, штат Нью-Йорк; Издательство Оксфордского университета; 1990: 469-471.
СТАТЬИ В ЖУРНАЛЕ
Canman CE, et al. Активация киназы АТМ ионизирующим излучением и фосфорилированием р53. Наука. 1998; 281: 1677-1679.
Банин С. и др. Усиленное фосфорилирование p53 ATM в ответ на повреждение ДНК. Наука. 1998; 281: 1674-1677.
Вт GD, et al. Построение карты транскрипции вокруг гена атаксии телеангиэктазии: идентификация по крайней мере четырех новых генов.Геномика. 1997; 40: 267-276.
Юнг М. и др. Регуляция p53 в ответ на ионизирующее излучение в фибробластах при атаксии телеангиэктазии. Int J Radiat Oncol Biol Phys. 1997; 37: 417-422.
Дорк Т. и др. Частый полиморфизм гена, мутировавшего при атаксии и телеангиэктазии. Зонды Mol Cell. 1997; 11: 71-73.
Fritz E, et al. Сверхэкспрессия усеченной топоизомеразы III человека частично корректирует множественные аспекты фенотипа атаксии-телеангиэктазии. Proc Natl Acad Sci USA.1997; 94: 4538-4542.
Савицкий К и др. Один ген атаксии телеангиэктазии с продуктом, подобным киназе PI-3. Наука. 1995; 268: 1749-1753.
Swift M и др. Заболеваемость раком в семьях с атаксией и телеангиэктазией. New Engl J Med. 1991; 325: 1831-1836.
Conerly SL, et al. Атаксия-телеангиэктазия или синдром Луи-Бар. J Am Acad Dermatol. 1985; 12; 681-696.
Дули Д.М. и др. Лечение пациентов с дегенеративными заболеваниями центральной нервной системы электростимуляцией спинного мозга.Confin Neurol. 1981; 44; 71-76.
ИЗ ИНТЕРНЕТА
Онлайн-Менделирующее наследование в человеке, OMIM (TM). Университет Джона Хопкинса, Балтимор, Мэриленд. МИМ-номер 208900; 1/3/00. Доступно по адресу: http://www.ncbi.nlm.nih.gov/htbin-post/Omim/dispmim?208900.
Национальный институт неврологических заболеваний и Сток. Информационная страница об атаксии и телагиэктазии. 29 октября 2003 г. Доступно по адресу: http://www.ninds.nih.gov/health_and_medical/disorders/a-t.htm?format=printable
Ataxia Telangiectasia: информационный бюллетень — Национальный институт рака
http: // www.Cancer.gov/cancertopics/factsheet/ataxiaaqa
Слышали ли вы о неврологе, стоящем за синдромом Луи-Бара? …: Неврология сегодня
By Dawn Фаллик
19 марта 2020
Объяснение наукиКраткое описание статьи
Женщина, связанная с одноименным синдромом Луи-Бар, является предметом обсуждения — и награды — о проблемах, с которыми столкнулась женщина в неврологии из более ранней эпохи.
Доктор.Дениз Луи-Бар
Когда Элизабет А. Кун, доктор медицины, начала исследование Дениз Луи-Бар, доктора медицины, невролога, которая помогла определить одноименный детский неврологический синдром, доктор Кун не был уверен, что выбрала правильную тему.
Будучи взрослым неврологом, доктор Кун, доцент кафедры неврологии в клинике Мэйо в Рочестере, штат Миннесота, никогда не встречал человека с болезнью, которую помог обнаружить доктор Луис-Бар — атаксии телеангиэктазии. Но в тот же день в своей клинике она встретила джентльмена с семейной атаксией и глазной телеангиэктазией, симптомы которого очень похожи на симптомы синдрома Луи-Бар.
«Это должно было быть, — сказал доктор Кун.
Ее статья о докторе Луи-Бар была опубликована в журнале Neurology в 2018 году. Но в этом году доктор Кун получает премию AAN McHenry в истории за свое исследование невролога, стоящего за атаксией телеангиэктазией. Заболевание вызывается мутациями в гене ATM и проявляется как мультисистемное нейродегенеративное расстройство с ранним началом, характеризующееся прогрессирующим отсутствием координации, которое проявляется у малышей, когда они начинают учиться ходить.У пациентов также высока вероятность лейкемии или лимфомы.
Доктор Луи-Бар впервые описал заболевание в 1941 году в международном журнале Confinia Neurologica , опубликованном в Швейцарии. Ей тогда было 27 лет. Но, как отмечается в статье Neurology , ей никогда не удавалось дать название расстройству. Действительно, цитата в статье Neurology , взятой из литературы, подчеркивает, как в то время воспринималось положение женщины в медицине: «Доктор.Г-н Резник вспоминал Луи-Бар во время ее пребывания в Льеже: «Я действительно знал г-жу Луи-Бар, когда я, будучи молодой студенткой, работала старшим помощником в службе Брюллов. Ее можно было узнать по пышным рыжим волосам ». Как отмечалось в статье, доктор Луи-Бар стал« нейропсихиатром »в отделении внутренней медицины вместе с известным физиологом профессором Люсьеном Брюллем в Государственном университете Льежа.
«Она явно была очень опытным врачом и исследователем, но в статьях о ней описывается ее внешний вид, особенно то, что у нее были« пышные рыжие волосы ».’” —ДР. ЭЛИЗАБЕТ А. КУН
Доктор Кун обсудила с Neurology Today , что она узнала о докторе Луи-Бар и как это повлияло на ее роль как невролога сегодня. Отрывки из ее высказываний приводятся ниже.
В своем исследовании вы пишете о том, как доктор Луи-Бар заинтересовалась лечением детей, потому что она увидела несправедливость в отношении здоровья и проблемы со здоровьем у маленьких детей. Как вы заинтересовались неврологией?
Я интересовался медициной с подросткового возраста.Мой дядя занимался семейной медициной, и я знал, что хочу заботиться о людях, а также заниматься научной и серьезной деятельностью. Мой интерес к неврологии возник из личного опыта. Я помню, как был студентом, я работал в исследовательской лаборатории, изучая последовательности ДНК у рыбок данио, у которых были неврологические расстройства. Я также следил за Хэнком Полсоном [Генри Полсон, MD, PhD], который ухаживал за пациентами с болезнью Хантингтона в многопрофильной клинике. Итак, утром я смотрел на генетический код, а днем я видел, как пациенты сталкиваются со значительными симптомами, вызванными мутациями в их генетическом коде.
Что вас удивило в вашем исследовании доктора Луи-Бара?
Я знала об этой болезни, атаксии и телеангиэктазии, но не знала, кто она такая. Сначала я подумал, что болезнь назван в честь двух разных людей, и решил, что это мужчины. Но по-настоящему меня заинтересовало то, как болезнь получила ее имя. На самом деле она была не первым, кто описал эту болезнь — это были два чешских врача в 1926 году, но они описали ее как «расстройство типа дистонии», и поэтому это не было связано с Луи-Баром.
Группа, которая действительно больше всего работала над атаксией-телеангиэктазией, была из Калифорнии в середине 20-го века под руководством доктора Елены Бодер, американского детского невролога. Были некоторые разногласия по поводу названия болезни «синдром Луи-Бар» по сравнению с атаксией телеангиэктазией, потому что о ней писали другие люди, и была некоторая проблема с патофизиологическим описанием АТ доктором Луи-Баром. Но если посмотреть литературу, ее имя все еще часто используется.
Считаете ли вы, что синдром не следует называть в честь доктора Х.Луи-Бар?
Меня это не особо беспокоит, поскольку Елена Бодер значительно продвинулась в этой области, и я думаю, что атаксия телеангиэктазия — хорошее описательное название.
Что исследование показало, как к доктору Луи-Бар относились в этой области?
Читая между строк в историческом сочинении, вы понимаете, что она не была полностью частью медицинского сообщества того времени. После статьи в журнале, которую она написала в 1941 году, последовало продолжение ее коллег и наставника, и они даже не использовали ее имя; не было описания ее работы.Я чувствую, что, возможно, у нее не было всей поддержки, которую она могла бы использовать.
Были ли еще какие-нибудь подсказки о том, что значит быть женщиной в медицине в то время?
Она явно была очень опытным врачом и исследователем, но в статьях о ней описывается ее внешний вид, особенно то, что у нее были «пышные рыжие волосы». Эти физические характеристики беспокоили меня, поскольку мы знаем, что женщин чаще судят по внешнему виду. Она не осталась в академии; ее муж уехал в Бельгию, и она покинула академический мир, когда переехала.Она продолжала практиковать и открыла специализированные клиники по уходу за детьми и взрослыми с ограниченными возможностями, что благородно.
Что еще вы извлекли из своего исследования о ней?
Когда она училась, это было очень продуктивное время для нее. Позже она посвятила свое время многопрофильной помощи, особенно детям с умственными недостатками, и проделала потрясающую работу в этом отношении. Она была очень внимательным клиницистом и действительно много работала в области внутренней медицины и неврологических расстройств, уделяя особое внимание уходу за пациентами, что продемонстрировало ее разнообразные способности и интересы.
Как вы соотносите историю доктора Луи-Бар с вашим опытом работы в неврологии сегодня?
Это прекрасное время, чтобы быть женщиной в медицине. Женщины все чаще занимают руководящие должности, и я благодарна за то, что работаю в отделе, возглавляемом женщиной, доктором медицины Клаудией Лучинетти. Очень важно найти наставников, которые поддержат вас и помогут сориентироваться в вашей карьере, и я также благодарен своим многочисленным наставникам. Для женщины на пути будут возникать микроагрессии и препятствия, но найти наставников, которые помогут вам управлять и продолжать двигаться вперед, очень важно.
Как исследование повлияло на ваше мнение о работе с пациентами с двигательными расстройствами?
Я бы сказал, что пациенты приходят с самыми разными симптомами и происхождением. Все пациенты нуждаются в помощи по-разному. Вы должны научиться адаптировать свой подход к пациенту. Это не просто знание того, что у пациента конкретный тип заболевания; Речь идет о необходимости думать о том, как болезнь влияет на конкретного пациента, как симптомы влияют на все аспекты его жизни.Меня учили, что это в ординатуре по цитате сэра Уильямса Ослера: «Намного важнее знать, какой тип пациента болен, чем какой у него заболевание».
Раскрытие информации
Доктор Кун не раскрывала информацию .
Телеангиэктазия — Детская исследовательская больница Св. Иуды
Что такое атаксия-телеангиэктазия?
Атаксия-телеангиэктазия — редкое генетическое заболевание, поражающее нервную систему, иммунную систему и другие системы организма.У детей с этим заболеванием наблюдается атаксия или проблемы с координацией движений. У них также есть небольшие скопления увеличенных кровеносных сосудов, называемые телеангиэктазиями, которые возникают в глазах и на поверхности кожи. По сравнению с людьми, у которых нет атаксии-телеангиэктазии, они подвержены более высокому риску развития определенных типов рака крови и рака иммунной системы.
У людей с атаксией-телеангиэктазией атаксия часто начинается в раннем детстве, обычно в возрасте до 5 лет, и со временем ухудшается.Большинству детей с атаксией-телеангиэктазией в конечном итоге становится трудно ходить, и у них возникают проблемы с равновесием и координацией рук. У них могут быть и другие проблемы с нервной системой, в том числе следующие:
- Невнятная речь
- Глазодвигательная апраксия (затруднение движения глазами из стороны в сторону)
- Хорея (непроизвольные рывки)
- Миоклонус (мышечные подергивания)
- Невропатия (нарушение функции нервов).
К подростковому возрасту многим людям с атаксией-телеангиэктазией потребуется инвалидная коляска.
Что вызывает атаксию-телеангиэктазию?
Атаксия-телеангиэктазия вызывается изменениями в гене, известном как ATM . Гены несут информацию, сообщающую клеткам в теле, как функционировать. Ген ATM необходим клеткам для восстановления поврежденного генетического материала (ДНК).
Большинство людей без атаксии-телеангиэктазии несут в своих клетках две рабочие копии гена ATM . Один экземпляр наследуется от матери, а другой — от отца. Клетки людей с атаксией-телеангиэктазией несут в своих клетках две измененные копии гена ATM .Одна измененная копия была унаследована от матери, а другая измененная копия была унаследована от отца. Изменения вызывают некорректную работу гена ATM . Их называют мутациями.
Когда обе копии гена ATM имеют мутации, клетки не могут восстанавливать повреждения своей ДНК так же, как и те, у кого нет атаксии-телеангиэктазии. Клетки становятся нестабильными и иногда умирают или разрастаются, образуя опухоль. Вот почему люди с атаксией-телеангиэктазией подвержены более высокому риску развития некоторых видов рака.
Люди с только одной мутированной копией гена ATM не страдают атаксией-телеангиэктазией и не проявляют признаков и симптомов этого состояния. Этих людей называют «носителями». Женщины-носители (женщины с одной мутированной копией гена ATM ) имеют немного более высокий риск развития рака груди, чем население в целом.
родителей ребенка с атаксией-телеангиэктазией имеют 25% (1 из 4) шанс родить еще одного ребенка с этим заболеванием.У них есть 50% (1 из 2) шанс иметь ребенка, у которого будет только одна копия мутации ATM . Этот ребенок будет носителем. Носители не страдают атаксией-телеангиэктазией, но могут передавать мутировавший ген ATM своим собственным детям. Вероятность рождения еще одного ребенка с двумя нормальными копиями гена ATM составляет 25% (1 из 4). У этого ребенка не будет атаксии-телеангиэктазии и он не будет носителем. Дети с двумя нормальными копиями гена ATM передадут своим собственным детям только нормальные копии гена ATM .
A Человек с атаксией-телеангиэктазией имеет различные риски рождения ребенка с атаксией-телеангиэктазией. В частности, человек с атаксией-телеангиэктазией передаст по одной мутации ATM каждому из своих детей. Вероятность того, что у этого человека родится ребенок с атаксией-телеангиэктазией, зависит от того, несет ли его или ее партнер также мутацию ATM . Если партнер является носителем мутации ATM , вероятность рождения ребенка с этим заболеванием составляет 50% (1 из 2).Вероятность рождения ребенка, у которого будет только одна копия мутации ATM , составляет 50% (1 из 2). Если у партнера нет мутации ATM , у детей не будет атаксии-телеангиэктазии, но все дети будут носителями.
Каков риск рака у людей с атаксией-телеангиэктазией?
У людей с атаксией-телеангиэктазией вероятность развития рака составляет около 40%. Из тех, у кого развивается рак, у большинства разовьется лейкемия (рак клеток крови) или лимфома (рак иммунных клеток).Другие типы рака, которые могут развиться, включают следующие:
- Рак яичников
- Рак груди
- Рак щитовидной железы
- Опухоли околоушной (слюнной) железы
- Рак желудка (желудка)
- Меланома (рак кожи)
- Леи опухоли гладких мышц)
Женщины-носители (женщины с одной мутированной копией гена ATM ) имеют немного более высокий риск развития рака груди, чем те, кто не являются носителями.Риск относительно низкий (примерно в 2–4 раза выше, чем у населения в целом). Учитывая этот риск, некоторые медицинские работники могут рекомендовать женщинам-носителям мутации ATM рассмотреть возможность скрининга рака груди в более раннем возрасте, чем людям, не являющимся носителями. В настоящее время считается, что носители мутации ATM не подвержены повышенному риску других типов рака, кроме рака груди.
Какие еще физические признаки наблюдаются у людей с атаксией-телеангиэктазией?
Люди с атаксией-телеангиэктазией могут иметь другие проблемы со здоровьем.Не все люди с атаксией-телеангиэктазией будут иметь все перечисленные ниже физические признаки. Эти физические данные варьируются от человека к человеку.
- Ослабленная иммунная система, чаще приводящая к инфекциям
- Чувствительность к ионизирующему излучению (например, излучению, используемому для компьютерной томографии и медицинских рентгеновских лучей)
- Преждевременное старение с поседением волос
- Нарушение эндокринной (гормональной) системы, включая диабет 2 типа, низкий рост и задержку полового созревания
- Легкие проблемы с обучением или легкая умственная отсталость
- Мозжечок меньше нормы (часть мозга), который можно увидеть на МРТ после возраста примерно 7 или 8 лет
- Укороченная продолжительность жизни (большинство пациентов доживают до 20 лет; некоторые пациенты доживают до 40 или 50 лет)
Степень тяжести признаков и симптомов атакси-телеангиэктазии может варьироваться в зависимости от семьи.Однако люди с атаксией-телеангиэктазией, которые находятся в одной семье, часто имеют сходное течение болезни.
Как люди с атаксией-телеангиэктазией проверяются на наличие опухолей?
Людей с атаксией-телеангиэктазией должен лечить врач, который хорошо знает это состояние. Детям с атаксией-телеангиэктазией следует регулярно проходить обследования для выявления рака как можно раньше. Цель скрининга — выявление и раннее лечение рака, чтобы обеспечить наилучший результат для пациентов.
В настоящее время нет установленных руководств по скринингу на рак у детей с атаксией-телеангиэктазией. Обычно рекомендуется следующее:
- Ежегодная лабораторная работа (общий анализ крови, полная метаболическая панель и лактатдегидрогеназа) для выявления лимфомы и лейкемии.
- Ежегодный медицинский осмотр врачом, который хорошо знает это состояние, уделяя особое внимание любым признакам лейкемии или лимфомы (потеря веса, синяки и локализованная боль или опухоль).
- Ежегодные осмотры у педиатра.
Для носителей одной копии мутировавшего гена ATM может быть рекомендован ранний скрининг рака груди. Это должен решать врач, который ведет пациентов с повышенным риском рака груди.
Если рак обнаружен, очень важно найти врача, имеющего опыт лечения рака у людей с атаксией-телеангиэктазией. Стандартные дозы некоторых видов химиотерапии и лучевой терапии могут быть более вредными для пациентов с атаксией-телеангиэктазией.Могут потребоваться более низкие дозы химиотерапии или лучевой терапии.
Возможно, рекомендуемые обследования со временем могут измениться по мере того, как врачи узнают больше об атаксии-телеангиэктазии. Родители должны обсудить все варианты скрининга своего ребенка с врачом, который хорошо знает это состояние. Поскольку атаксия-телеангиэктазия — сложное заболевание, очень важно, чтобы родители обратились к опытному врачу для своего ребенка.
Как диагностируется атаксия-телеангиэктазия?
Врач может заподозрить атаксию-телеангиэктазию на основании признаков и симптомов этого состояния.Для подтверждения клинического диагноза атаксия-телеангиэктазия можно использовать несколько лабораторных тестов:
- Специализированные тесты для измерения количества белка ATM в клетках. Ген ATM образует белок, также называемый ATM. Если у человека есть мутированная копия гена ATM , у этого человека обычно уровни белка ATM в клетках намного ниже нормального.
- Анализы крови для измерения уровня белка, называемого альфа-фетопротеином. Более 95% людей с атаксией-телеангиэктазией имеют высокий уровень альфа-фетопротеина, но у большинства людей без этого заболевания уровни очень низкие.Врачи не знают, почему у людей с этим заболеванием так высок уровень альфа-фетопротеина.
- Тест на радиочувствительность для измерения того, насколько хорошо клетка может восстанавливать поврежденную ДНК. Нормальные клетки, содержащие рабочий белок ATM, должны иметь возможность восстанавливать себя после воздействия радиации. Клетки людей с атаксией-телеангиэктазией не выживают нормально после воздействия радиации, потому что они также не восстанавливают ДНК.
- Хромосомный анализ для поиска транслокации, которая может произойти в клетках, обработанных определенным химическим веществом.Хромосомы — это часть клетки, которая содержит генетический материал (ДНК). Транслокация происходит, когда хромосомы ломаются и фрагменты неправильно прикрепляются к другим хромосомам.
Как проводится генетическое тестирование на атаксию-телеангиэктазию?
Врач может заподозрить атаксию-телеангиэктазию, изучив медицинский или семейный анамнез человека и проведя некоторые из лабораторных тестов, перечисленных выше. В большинстве случаев врач или генетический консультант задаст вопросы о здоровье человека и других членов семьи.
Генетический консультант или врач запишет, у каких членов семьи появились признаки и симптомы атаксии-телеангиэктазии, включая рак, и в каком возрасте они возникли. На основе этой информации они создадут генеалогическое древо. Врачи и консультанты-генетики изучат генеалогическое древо, чтобы выяснить, совпадают ли закономерности с тем, что можно увидеть при атаксии-телеангиэктазии. Если заболевание подозревается у ребенка или других членов семьи, врач или генетический консультант, скорее всего, порекомендует генетическое тестирование гена ATM .
Диагностическое генетическое тестирование
Если врач или консультант-генетик подозревает, что у человека есть атаксия-телеангиэктазия, диагностическое обследование может проводиться следующим образом:
- Берут образец крови.
- ДНК выделяют из клеток в образце. Гены человека состоят из ДНК.
- Обе копии гена ATM человека проверяются на возможные изменения. Специалист-генетик сравнивает две копии гена ATM человека с нормальными копиями этого гена.Если есть различия, специалист решает, могут ли они вызвать определенное состояние, такое как атаксия-телеангиэктазия.
- Если мутация обнаружена в гене ATM , генетический консультант будет работать с семьей следующим образом:
- Чтобы помочь семье понять риски атаксии-телеангиэктазии
- Чтобы выяснить, следует ли другим членам семьи рассмотреть возможность тестирования на мутацию
- Чтобы помочь в принятии решений о пренатальном генетическом тестировании
Важно помнить, что генетическое тестирование не всегда обнаруживает мутации в гене ATM у всех людей с атаксией-телеангиэктазией.У человека все еще может быть атаксия-телеангиэктазия, даже если мутация ATM не обнаружена. Могут быть и другие гены, связанные с заболеванием, о котором врачи еще не знают, или могут быть мутации, которые тест не обнаружил.
Пренатальное генетическое тестирование
Родители могут пройти дородовое тестирование, чтобы выяснить, влияет ли беременность на известную в семье мутацию ATM . Тестирование может проводиться либо до наступления беременности, либо во время беременности.
Людям, рассматривающим возможность пренатального тестирования, следует обратиться к консультанту по генетическим вопросам, чтобы обсудить плюсы и минусы тестирования. Консультант-генетик также может помочь родителям подумать о том, как они хотят поступить с результатами тестирования.
Тестирование, проводимое до беременности — Тестирование, проводимое до беременности, называется предимплантационным генетическим тестированием (ПГТ). Этот особый тип генетического тестирования проводится вместе с экстракорпоральным оплодотворением (ЭКО). PGT предлагает способ тестирования эмбрионов на наличие известных мутаций ATM перед их помещением в матку.
Тестирование, проводимое во время беременности — Тестирование можно использовать, чтобы определить, влияет ли беременность на известную мутацию ATM . Врач собирает клетки беременных одним из двух способов:
- Взятие пробы ворсин хориона (CVS) — в течение первого триместра (первые три месяца)
- Амниоцентез — во втором триместре или позже (последние шесть месяцев). )
Собранную ткань можно проверить на наличие ATM мутаций, идентифицированных в семействе.
Оба этих теста несут незначительный риск и должны быть обсуждены с опытным врачом или генетическим консультантом.
Особые опасения
Генетическое тестирование на атаксию-телеангиэктазию — сложный процесс. Тем, кто думает о тестировании, следует подумать о преимуществах и рисках. Им следует обсудить этот процесс с генетическим консультантом до проведения тестирования. Если они выберут тестирование, им следует обсудить результаты теста с врачом или генетическим консультантом, чтобы убедиться, что они понимают значение результатов.
Иногда дети или взрослые с атаксией-телеангиэктазией могут чувствовать грусть, тревогу или гнев. Родители, которые передают мутацию ATM одному или нескольким своим детям, могут чувствовать себя виноватыми. У некоторых людей с мутацией ATM или мутациями в некоторых штатах могут возникнуть проблемы с получением страхового покрытия по инвалидности, страхования жизни или долгосрочного ухода. Более подробную информацию о генетической дискриминации можно найти на сайте www.ginahelp.org.
Требуются ли другие особые медицинские услуги для детей с атаксией-телеангиэктазией?
Из-за других медицинских проблем, связанных с атаксией-телеангиэктазией, другие специалисты должны осмотреть пациента:
- Неврология
- Иммунология
- Поддерживающая терапия (например, физиотерапия, трудотерапия и логопедия)
- Регулярные проверки развития и школы прогресс, как считают врачи, необходим ребенку.
Люди любого возраста с атаксией-телеангиэктазией имеют более высокий риск рака. Им следует следить за своим здоровьем и придерживаться здоровых привычек на протяжении всей жизни. Важно продолжать регулярные медицинские осмотры и осмотры. Таким образом, любой рак может быть обнаружен на самой ранней стадии, поддающейся лечению.
И люди с атаксией-телеангиэктазией, и носители более чувствительны к воздействию радиации. Это может увеличить риск развития рака. Таким образом, по возможности следует избегать медицинских обследований, в которых используется излучение, таких как компьютерная томография, рентген и маммография.Иногда врач может решить, что эти тесты необходимы, если польза от теста превышает риск развития рака в результате теста. Кроме того, людям с атаксией-телеангиэктазией требуется особая осторожность, когда требуется анестезия.
Другие идеи по снижению риска рака:
- Соблюдайте здоровую диету с большим количеством фруктов и овощей
- Регулярно выполняйте физические упражнения
- Избегайте курения или употребления табачных изделий
- Избегайте пассивного курения
- Избегайте чрезмерного пребывания на солнце и всегда на солнце носите солнцезащитный крем, головной убор и защитную одежду
Людям с атаксией-телеангиэктазией следует внимательно следить за общими признаками или симптомами, которые могут свидетельствовать о раке:
- Необъяснимая потеря веса
- Потеря аппетита
- Легкие синяки
- Кровь в стуле или изменения в кишечнике
- Боли, боли, уплотнения или припухлость, которые не могут быть объяснены
- Головные боли или изменения зрения или функции нервов, которые не проходят
В любом случае важно обратиться за медицинской помощью появляется необычное.
Обновлено 5/2020
Атаксия с телеангиэктазией. Информация о синдроме Луи-Бар. Пациент
Синоним: синдром Луи-Бара
Атаксия-телеангиэктазия (АТ) — редкое аутосомно-рецессивное заболевание. [1] Ответственный ген был локализован в 11q22.3-23.1. [2] A-T характеризуется: [3, 4, 5]
- Прогрессирующая нейродегенерация.
- Высокий риск злокачественных новообразований, особенно рака груди у женщин.
- Иммунодефицит.
- Повышенная чувствительность к ионизирующему излучению.
- Хромосомный разрыв (обычно t (7; 14) транслокации).
Существует значительная фенотипическая гетерогенность как в клинической картине (особенно в восприимчивости к легочной инфекции, наличии и степени когнитивных нарушений и предрасположенности к лейкемии), так и в скорости прогрессирования, что отражает аллельное разнообразие. [6]
Эпидемиология
A-T — редкое заболевание:
- Заболевание наследуется как аутосомно-рецессивный признак с полной пенетрантностью.Об этом сообщили во всем мире.
- Иногда это связано с высоким уровнем кровного родства. [7]
Носители (гетерозиготы) мутаций локуса А-Т имеют значительно меньшую ожидаемую продолжительность жизни по сравнению с лицами, не являющимися носителями, с повышенным риском смерти от рака и ишемической болезни сердца. Гетерозиготы женского пола имеют особый риск рака груди. [5, 8] Другие исследования поставили под сомнение эти результаты и, хотя они также подтвердили умеренный риск рака груди у гетерозигот A-T, не обнаружили больших специфических для мутаций различий в общем риске. [9]
Генетика
Молекулярная патология AT была хорошо описана в последнее время:
- Классическая форма AT возникает в результате присутствия двух мутантных генов AT (ATM) на хромосоме 11, что приводит к общему количеству потеря белка ATM (протеинкиназы). Белок ATM обычно распознает повреждение ДНК и активирует механизм восстановления ДНК и контрольные точки клеточного цикла, чтобы восстановить и минимизировать риск генетического повреждения.
- Понимание функции ATM через эффект потери A-T дало более широкое понимание предрасположенности к раку и некоторых аспектов нейродегенерации. [10]
- Более мягкие формы A-T с более поздним началом или медленным прогрессированием неврологической дегенерации, по-видимому, отражают некоторую сохраненную активность протеинкиназы ATM из-за воздействия различных мутаций. [11]
Презентация
[12]- A-T обычно проявляется в возрасте 2 лет с прогрессирующей мозжечковой атаксией.
- Другие симптомы, которые могут присутствовать, включают:
- Отсутствие выражения лица.
- Склонность к слюноотделению.
- Медленная невнятная речь.
- Рецидивирующие инфекции, особенно носовые и респираторные — из-за снижения уровня иммуноглобулина А (IgA).
- Телеангиэктазии развиваются примерно в возрасте 3 лет и могут отсутствовать до 10 лет, но будут присутствовать во всех случаях. Телеангиэктазии могут присутствовать на любом участке тела — чаще всего на конъюнктивах, ушных раковинах, лице, грудины и изгибах.
- Другие признаки включают:
- Атаксия туловища
- Глазодвигательная апраксия
- Снижение сухожильных рефлексов
- Легочные инфекции
- Инфекции синуса
- Бульбарная дисфункция
- Рецидивирующая аспирация 9024 Дистилляция 9024 9024 атония 9024
- Ограничение роста
Дифференциальный диагноз
Существует необходимость дифференцировать от других аутосомно-рецессивных атаксий: [13] атаксия Фридрейха (наиболее распространенная в европейских странах) и гораздо более редкие состояния, такие как атаксия с дефицитом витамина E, абеталипопротеинемия, болезнь Рефсума, спастическая атаксия, спиноцеребеллярная атаксия с младенческим началом и атаксия с глазодвигательной апраксией.
A-T обычно отличается более ранним возрастом начала, наличием телеангиэктазий и более поздним развитием дистонии и хореи, а также специальными исследованиями.
Исследования
[14]- Уровень альфа-фетопротеина в сыворотке (повышен на 90%).
- Генетическое тестирование генов ATM и отсутствие белка ATM в ядерных экстрактах.
- Тест на радиочувствительность in vitro (анализ выживаемости колоний).
Сопутствующие заболевания
Повышенный риск рака
- При вскрытии почти у половины людей обнаруживается злокачественная опухоль.
- Лимфоретикулярные злокачественные новообразования (лимфомы и острый лимфобластный лейкоз (ОЛЛ)) преобладают в первые два десятилетия, после чего появляются солидные опухоли.
Иммунодефицит
[15]- A-T ассоциируется с высокой распространенностью лабораторных иммунологических аномалий — чаще всего, дефицита IgA, IgG и IgE и лимфопении с уменьшением B-лимфоцитов и уменьшением CD4 и CD8 T-клеток.
- Рецидивирующие инфекции верхних и нижних дыхательных путей являются обычным явлением.Тяжелые системные бактериальные или вирусные и условно-патогенные инфекции при А-Т встречаются нечасто. Иммунный дефект не прогрессирует.
Ведение
В настоящее время нет лекарства и лечения для замедления прогрессирования атаксии. A-T — это мультисистемное заболевание, требующее множественных терапевтических вмешательств для замедления или остановки нейродегенерации, предотвращения или лечения опухолей и коррекции связанного с ними иммунодефицита. Несмотря на возросшее молекулярное понимание болезни, у нас нет эффективного ответа на эти проблемы.В будущем перенос эмбриональных стволовых клеток, использование антиоксидантов и таргетная генная терапия могут дать некоторую надежду. [6]
Прагматическое лечение этого состояния состоит из следующего:
- Неврологические симптомы трудно поддаются лечению. Лечение дисфункции базальных ганглиев может быть предпринято с помощью производных L-DOPA, антагонистов дофамина и холинолитиков. Потеря равновесия, проблемы с речью и координацией могут быть улучшены с помощью амантадина, флуоксетина и буспирона.Тремор можно лечить габапентином, клоназепамом и пропранололом. [6]
- Оперативное лечение инфекций и, в некоторых случаях, профилактическое использование антибиотиков. В некоторых центрах используются регулярные инъекции иммуноглобулинов. Эффективность имплантатов тимуса плода не доказана.
- Скрининг проблем, связанных с глотанием, и аспирации. Рассмотрите возможность использования загустителей и энтерального питания. [6]
- Некоторые выступают за регулярный скрининг злокачественных новообразований с помощью онкомаркеров FBC и сыворотки крови.
- Участие мультидисциплинарной команды — особенно важно участие физиотерапевтов, логопедов и эрготерапевтов.
- Режимы приема высоких доз витаминов, фолиевой кислоты и альфа-липоевой кислоты были поддержаны из-за их заявленных противораковых свойств. Однако поливитамины не устраняют атаксию A-T. [6]
- По возможности избегайте рентгеновских лучей, если только лечение не зависит от них и альтернатива (например, МРТ или УЗИ) невозможна.
Учтите повышенный риск для взрослых членов семей с А-Т рака.Рассмотрите возможность направления к специалистам по генетике и более раннего участия в программах скрининга.
Прогноз
- A-T — неизлечимая и неизлечимая болезнь.
- Большинство детей A-T в подростковом возрасте становятся инвалидами, хотя существуют и более легкие варианты.
- Общение становится все труднее по мере того, как ухудшается контроль над почерком, речью и движением глаз.
- Средняя выживаемость составляет 19-25 лет (широкий диапазон) со смертью от рака и дыхательной недостаточности. [16]
Профилактика
Пренатальное тестирование доступно для семей, относящихся к группе риска, но не проводится в плановом порядке.Ген ATM слишком велик, чтобы можно было провести практический мутационный анализ для клинического скрининга. [10]
Дениз Луи-Бар • LITFL • Медицинская библиотека эпонимов
Дениз Луи-Бар (1914-1999) была бельгийским психоневрологом.
В Брюсселе Луи-Бар работал психоневрологом, помогая людям с умственными недостатками. Она внесла большой вклад в создание специальных учреждений по уходу за ними и в развитие методов их медицинского и социального управления.
Ее одноименно помнят за описание атаксии-телеангиэктазии [синдрома Луи-Бара] (1941).
Биография
- 1914 — Родилась Дениз Бар 3 апреля в Льеже, Бельгия. До 10 лет жила с семьей в Испании.
- 1939 — доктор медицины, получив совместную степень по физическому воспитанию в Свободном университете Брюсселя. Вышла замуж за инженера-строителя Ф. Луи 8 августа, приняв фамилию Луи-Бар, расставленную через дефис,
- 1940 — резидент в Институте неврологии Бунге, Антверпен, с невропатологом Людо ван Богартом (1897-1989)
- 1943 — преподаватель фармакологии, Льежский университет
- 1945 — нейропсихиатр, Льежский университет
- 1957 — переехал в Брюссель, чтобы работать психиатром, с особым интересом к уходу за людьми с ограниченными интеллектуальными возможностями.
- 1999 — умер 2 ноября в Брюсселе
Медицинские эпонимы
Синдром Луи-Бара (1941) [также известный как атаксия-телеангиэктазия]
Синдром Луи-Бар — аутосомно-рецессивное мультисистемное нейродегенеративное заболевание с ранним началом, характеризующееся прогрессирующей мозжечковой атаксией, телеангиэктазией, иммунными дефектами и предрасположенностью к злокачественным новообразованиям. Пациенты обычно поступают в раннем детстве с прогрессирующей мозжечковой атаксией, а позже у них развиваются телеангиэктазии конъюнктивы, прогрессирующая неврологическая дегенерация, синопульмональные инфекции и злокачественные новообразования.Раннее проявление атаксии может быть ошибочно диагностировано как атаксический церебральный паралич до появления глазокожных телеангиэктазий, которые обычно развиваются в возрасте от 3 до 5 лет.
1926 — Первое описание было сообщено чешскими врачами Ладиславом Силлаба (1868-1930) и Камилом Хеннером (1895-1967) в их статье Contribution a l’indépendance de l’athétose double idiopathique et congénitale, опубликованной в Revue Neurologique. В отчете задокументировано 3 брата и сестры подросткового возраста с прогрессирующим хореоатетозом; глазные телеангиэктазии; и они признали, что расстройство было семейным.
1940 — В амбулаторной клинике Института Бунге (Антверпен) Луи-Бар обследовал девятилетнюю девочку, у которой в раннем детстве развились прогрессирующая атаксия, умственная отсталость и телеангиэктазия. Луи-Бар опубликовала свой отчет в Confinia Neurologica в 1941 г. О прогрессирующем синдроме, включающем кожные капилляры и симметричные конъюнктивальные телеангиоэктазии, диспозицию кожных капилляров и симметричных конъюнктивальных телеангиоэктазий, дисфункция]
Автор описывает случай 9-летнего ребенка с синдромом мозжечка с преобладанием атаксии-абазии с нарушением речи и умственной отсталостью, прогрессирующим с третьего года жизни.Что касается кожи, то у ребенка регулярно и симметрично расположены пятна телеангиэктазий (отличных от простой ангиомы), а также систематически упорядоченные пятна «с молоком», напоминающие папоротник. Двойное состояние в отношении кожи и нервов позволяет разместить этот футляр в целом в сфере факоматозы без придания ему какой-либо конкретной формы
Louis-Bar 1941: 41
1957 — Елена Бодер (1908-1995), российско-американский детский невролог и Роберт Седжвик, представили предварительный отчет о 7 случаях и в 1958 году опубликовали серию из 8 случаев атаксии-телеангиэктазии, связанных с наследственным компонентом заболевания. .Boder и Sedgewick предложили термин «атаксия-телеангиэктазия» для обозначения семейного синдрома прогрессирующей мозжечковой атаксии, окулокожных телеангиэктазий и частых легочных инфекций. Они признали «пророческий клинический отчет об одном несемейном случае без вскрытия, опубликованный Луи-Баром в 1941 году» и признали, что первоначальное описание содержало два основных компонента синдрома. Тем не менее, авторы по-прежнему утверждали, что они изолировали «относительно новый синдром, который стал самостоятельным субъектом только в течение последних пяти лет».
1958 — Одноименный термин «синдром Луи-Бара» был впервые предложен Уиллардом Центруолл и Мелбой Миллер в публикации, касающейся двух пациентов с атаксией-телеангиэктазией. [AMA J Dis Child 1958]
… определенные синдромы, затрагивающие кожу, глаза и центральную нервную систему, например цефалоретинальный ангиоматоз (болезнь фон Гиппеля-Линдау), церебральный ангиоматоз (синдром Стерджа-Вебера), нейрофиброматоз (болезнь фон Реклингхаузена) и туберозный склероз (болезнь Бурнвилля) ).Следуя этим примерам маркировки патологией и собственными именами, было бы разумно назвать этот синдром «цефало-окулокутанной телеангиэктазией» (синдром Луи-Бара) . Интересно отметить, что Людо ван Богарт, который в 1940 году сообщил о первом случае «цефало-окулокутанной телеангиэктазии» мадам Луи-Бар, является тем же человеком, который пятью годами ранее ввел термин нейроэктодермальная дисплазия.
Centerwall, Миллер 1958: 394
1964 — Однако споры по поводу номенклатуры продолжались … Миллер и Гуди утверждали, что мадам Луи-Бар сначала четко обозначила синдром, который позже получил более подробный набор описательных признаков.
По всем этим причинам, из признания блестящей изоляции клинической сущности, полноты, не закрывающей двери для решения проблемы, благозвучия и похвалы, где заслуживает похвалы, давайте согласимся, во всяком случае за время, чтобы обозначить это расстройство как «синдром Луи-Бар».
Миллер, Гуди 1964: 586
1967 — Хеннер представил рукопись, озаглавленную «По поводу описания« телеангиэктазии атаксии », сделанного мадам Луи-Бар.Приоритет описания, Лад. Силлаба и К. Хеннер в 1926 г. о сосудистой сети конъюнктивы ». Статья была опубликована в Revue Neurologique в 1968 году, через год после смерти Хеннера.
Атаксия-телеангиэктазия — это преобладающий термин, используемый для обозначения этого заболевания, однако одноименный термин «синдром Луи-Бар» все еще используется в медицинской литературе.
Основные публикации
Споры
Приоритет синдрома доказан. Первым описанием часто считается описание Силлабы и Хеннера (1926).
Список литературы
Биография
Эпоним
- Syllaba L, Хеннер К. Вклад в независимость латышской двойной идиопатической и конгениальной, Revue Neurologique 1926; 1: 541–562.
- Boder E, Sedgwick RP. Атаксия-телеангиэктазия; семейный синдром прогрессирующей мозжечковой атаксии, окулокожных телеангиэктазий и частых легочных инфекций. Педиатрия. 1958; 21 (4): 526-554.
- Centerwall WR, Миллер MM. Атаксия, телеангиэктазия и синопульмональные инфекции; синдром медленно прогрессирующего ухудшения в детстве.AMA J Dis Child. 1958; 95 (4): 385-396.
- Миллер С.Дж., Гудди В. Синдром мадам Луи-Бар; история болезни с комментариями к названию, классификации и значимости этого расстройства. Brain 1964; 87: 581–588
- Мартин Л. Ретроспективное исследование нозологической позиции атаксии-телеангиэктазии первого наблюдения. J Neurol Sci. 1966; 3 (1): 2-9.
- Хеннер К. Описание г-жи Луи-Бар де л ‘«Телеангиэктазия атаксии». Priorité de la description, par Lad.Syllaba et K. Henner en 1926, du réseau vasculaire cononctival [По поводу описания «Телеангиэктазии атаксии» г-жой Луи-Бар. Приоритет описания, Лад. Силлаба и К. Хеннер в 1926 г. о сосудистой сети конъюнктивы]. Revue Neurologique. 1968; 118 (1): 60-63.
- Бодер Э. Атаксия-телеангиэктазия: некоторые исторические, клинические и патологические наблюдения. Врожденные дефекты. Серия оригинальных статей 1975; 11: 255–270.
- СИНДРОМ ЛУИ-БАРА. OMIM 208900. OMIM
эпоним
лицо, стоящее за именем
В 2017 году окончил Кардиффскую медицинскую школу, получив степень бакалавра психологии и медицины.В настоящее время работает врачом в отделении неотложной помощи в больнице сэра Чарльза Гэрднера в Перте, Австралия.
Врач скорой помощи МА (Оксон) МБЧБ (Эдин) ФАЦЕМ ФФСЭМ со страстью к регби; история болезни; медицинское образование; и асинхронное обучение евангелист #FOAMed. Соучредитель и технический директор Life in the Fast lane | Эпонимы | Книги |
синдром Луи-Бар | Наследственные глазные болезни
Телеангиэктазы часто обнаруживаются в ушных раковинах, щеках и предплечьях, обычно после появления неврологических симптомов.Однако это также заболевание с множественными системными признаками, наиболее серьезными из которых являются необычная чувствительность к ионизирующему излучению, чрезмерный хромосомный разрыв, недостаточность иммунной системы, легкие когнитивные нарушения и повышенный риск злокачественных новообразований. Лимфомы, часто B-клеточного происхождения, и лейкоз, обычно T-клеточного происхождения, являются наиболее распространенными злокачественными новообразованиями, но также значительно повышается риск рака груди. Уровни IgG2 и IgA в сыворотке часто снижаются, часто встречаются синопульмональные инфекции.Уровни альфа-фетопротеина в сыворотке обычно повышены. Атаксия прогрессирует и часто начинается с неустойчивости туловища с последующим поражением конечностей. Это часто сопровождается хореоатетозом и / или дистонией, что может привести к тяжелой инвалидности ко второму десятилетию. Продолжительность жизни сокращается, и многие пациенты умирают от своей болезни к 3 -му и 4 -м десятилетиям.
В некоторых семьях с подтвержденными мутациями в ATM заболевание проявляется признаками первичной торсионной дистонии и миоклонической дистонии.Эти признаки могут напоминать очевидный аутосомно-доминантный образец передачи инфекции от родителей к детям. Неясно, представляют ли эти семьи вариант AT или уникальное заболевание. О последнем свидетельствует более раннее проявление симптомов, отсутствие атрофии мозжечка и отсутствие атаксии и глазных телеангиэктазий при первичном обращении. Риск злокачественных новообразований в этих семьях высок.
О некоторых из этих признаков в более легкой форме сообщалось и среди гетерозиготных носителей.Наиболее серьезным из них является повышенный риск злокачественных новообразований, возможно, в 6,1 раз больше, чем у лиц, не являющихся носителями.