Атаксия Фридрейха
Атаксия Фридрейха (АФ) — аутосомно-рецессивное заболевание, т.е. больные дети рождаются у пары родителей, которые оба клинически здоровы, но являются носителями патологического гена. Заболевание поражает нейроны центральной и периферической нервной системы: преимущественно поражаются пучки Голля, в меньшей степени пучки Бурдаха, Флексига, Говерса, а так же, пирамидные пути, задние корешки, спинномозговые ганглии и периферические нервы,клетки коры мозжечка, базальные ганглии, кора головного мозга, проводящие пути спинного мозга. В других системах заболеванию подвергаются не менее важные клетки органов, это клетки миокарда, β — клетки островков Лангерганца в поджелудочной железе, клетки сетчатки и костных тканей. Вызывает прогрессирующую дегенерацию центральной и периферической нервной системы. Большинство больных гомозиготны; содержание мРНК у них так мало, что иногда она вообще не определяется (в отличие от здоровых лиц и носителей гена атаксии Фридрейха).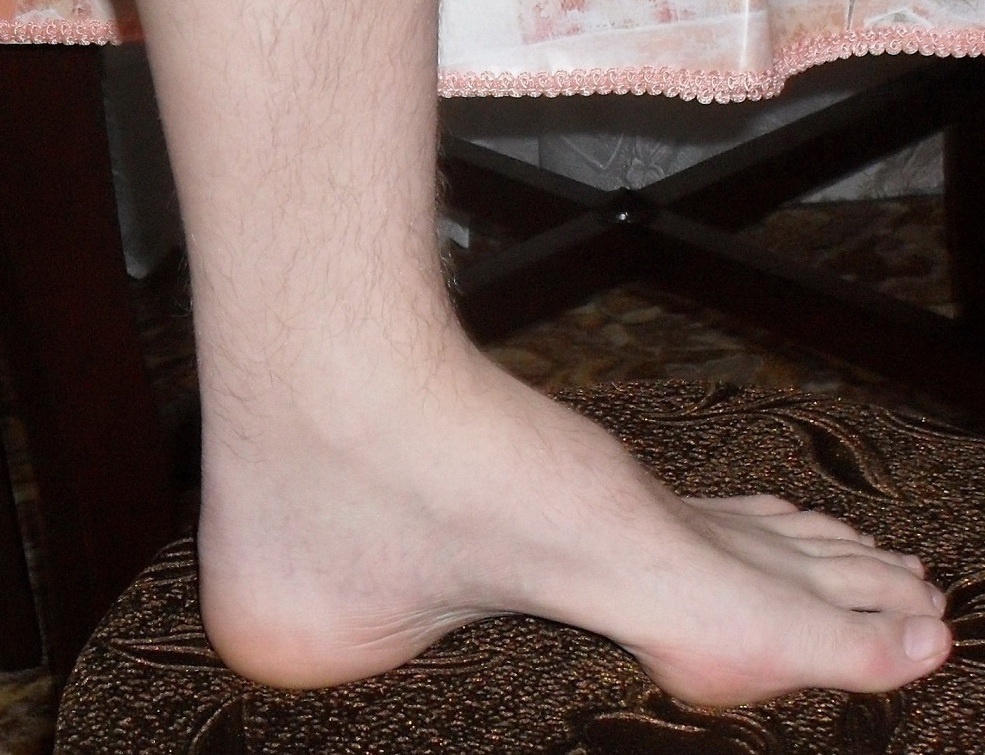
Симптомы атаксии Фридрейха чаще появляются на первом, втором десятилетии жизни, изредка на третьем и четвертом десятилетии. Появляется неуверенность, пошатывание, спотыкание при ходьбе, частые падения, нарушается почерк из-за тремора, появляется дизартрия, слабость в ногах, нарушается слух. Исчезают сухожильные и надкостничные рефлексы (в первую очередь ахиловые и коленные). Иногда ранним симптомом может быть ревмокардит. Больные не выполняют пяточно-коленную пробу, появляется покачивание в позе Ромберга, которое усиливается при закрывании глаз, расстройства сидения. Постепенно нарушается глубокая чувствительность, нарастает мышечная атрофия, на начальных этапах более выражена на нижних конечностях, с течением болезни захватывает и верхние. Формируется тотальная арефлексия. Атрофируется зрительный нерв, развивается катаракта, что ведет к слепоте, нарушается функция тазовых органов, развивается деменция. Развиваются эндокринные нарушения: сахарный диабет, гипогонадизм, инфантилизм, дисфункция яичников. Кардиомиопатии. Скелетные деформации: искривление позвоночника, кифосколиоз, «стопа Фридрейха» (высокий вогнутый свод стопы с переразгибанием пальцев в основных фалангах и сгибанием в дистальных), деформация пальцев рук и ног, косолапость. Течение болезни неуклонно прогрессирующее, при отсутствии адекватного лечения, длительность болезни обычно не превышает 20 лет. Непосредственной причиной смерти могут быть сердечная и легочная недостаточность, инфекционные осложнения. В редких случаях при отсутствии сахарного диабета и сердечных нарушений, больные доживают до 70-80 лет. |

Диагностика: Компьютерная томография головного мозга, которая остается основной диагностикой атаксий при этом заболевании малоэффективна, т.к. обнаруживает изменения только на поздних стадиях. Удается обнаружить только слабую степень атрофии мозжечка на ранней стадии и атрофию полушарий, расширение стволовых цистерн, боковых желудочков и субарахноидального пространства обоих полушарий на более поздних стадиях. Ранняя диагностика атаксии Фридрейха производится с помощью МР-томографии, которая дает возможность обнаружить атрофию спинного мозга и уменьшение поперечного размера спинного мозга, особенно усиливающееся в каудальном направлении на развернутой стадии, и умерено выраженную атрофию моста, мозжечка и продолговатого мозга. На начальной стадии обязательно проводится электрофизиологическое исследование, при таких исследованиях устанавливается тяжесть поражения чувствительности нервов конечностей.
Дифференцирование диагноза: Схожая симптоматика характерна для опухолей мозжечка, наследственных обменных заболеваний: Gm 1- и Gm 2-ганглиозидоз и галактосиалидоз (для дифференциации проводится исследование активности β-галактозидазы и гексозаминидазы А), нейросифилиса, фуникулярного миелоза, наследственной атаксии при дефиците витамина Е, синдрома Бассена-Корнцвейга, наследственных обменных заболеваниях, таких как болезнь Краббе (дифф.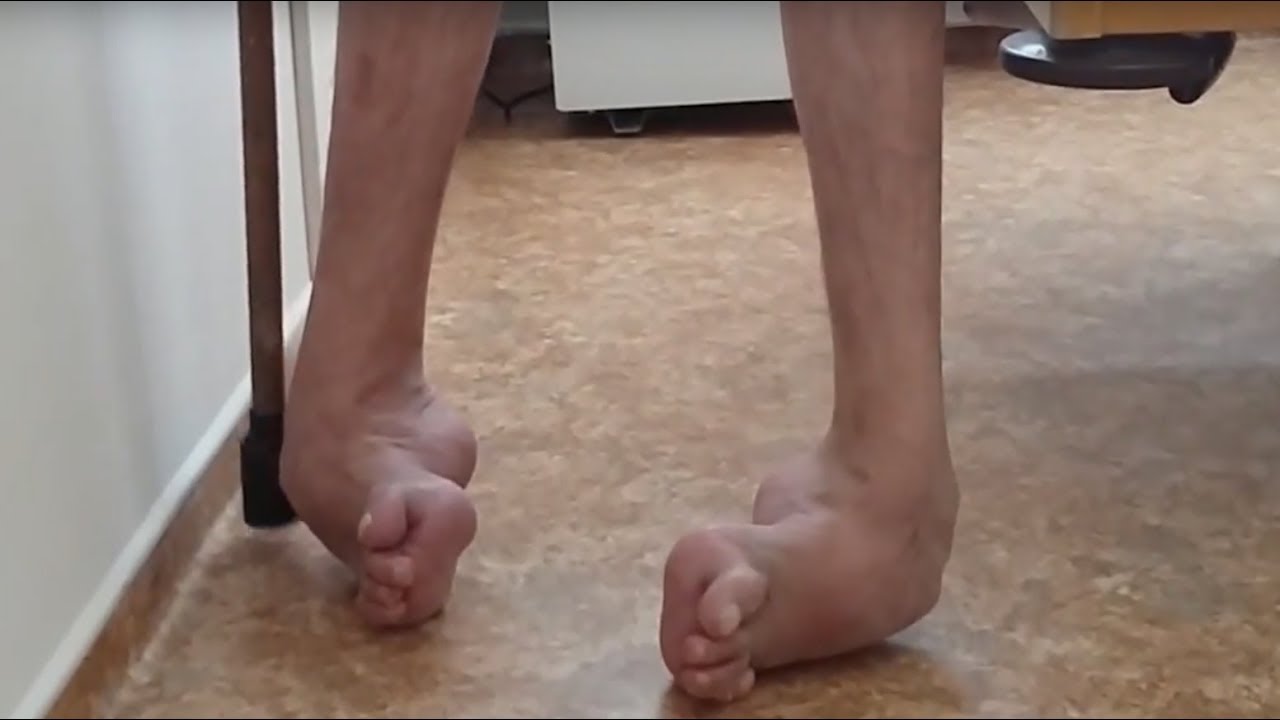

Лечение атаксии Фридрейха не приводит к полному выздоровлению, но своевременная профилактика дает возможность избежать развития многих симптомов и осложнений. Для замедления прогрессирования заболевания назначаются препараты митохондриального ряда, антиоксиданты и другие препараты, уменьшающие аккумуляцию железа в митохондриях. Общий принцип лечения данными препаратами состоит в сочетанном назначении лекарств, синергично влияющих на разные уровни энергетического метаболизма. Рекомендуется одновременное назначение как минимум трех лекарственных средств из первых трех групп. (Препараты, повышающие активность дыхательной цепи митохондрий, кофакторы энзимных реакций энергетического обмена, антиоксиданты).Назначаются такие антиоксиданты, как витамины А и Е, а также синтетический заменитель коэнзима Q 10 – идебенон, который затормаживает нейродегенеративный процесс и развитие гипертрофической кардиомиопатии. Обычно назначают препараты, улучшающие метаболизм миокарда: рибоксин, кокарбоксилазу, предуктал и др. Назначается также 5-гидроксипрофан, который дает, неплохие результаты, но требует дальнейших исследований. В целом лечение симптоматическое, направлено на такие симптомы, как сахарный диабет, заболевания сердечно-сосудистой системы. Проводится общеукрепляющее лечение (витамины), а также препараты, влияющие на тканевый обмен (пирацетам, аминалон, ацефен, церебролизин), лечение которыми следует периодически повторять.
Общий принцип лечения данными препаратами состоит в сочетанном назначении лекарств, синергично влияющих на разные уровни энергетического метаболизма. Рекомендуется одновременное назначение как минимум трех лекарственных средств из первых трех групп. (Препараты, повышающие активность дыхательной цепи митохондрий, кофакторы энзимных реакций энергетического обмена, антиоксиданты).Назначаются такие антиоксиданты, как витамины А и Е, а также синтетический заменитель коэнзима Q 10 – идебенон, который затормаживает нейродегенеративный процесс и развитие гипертрофической кардиомиопатии. Обычно назначают препараты, улучшающие метаболизм миокарда: рибоксин, кокарбоксилазу, предуктал и др. Назначается также 5-гидроксипрофан, который дает, неплохие результаты, но требует дальнейших исследований. В целом лечение симптоматическое, направлено на такие симптомы, как сахарный диабет, заболевания сердечно-сосудистой системы. Проводится общеукрепляющее лечение (витамины), а также препараты, влияющие на тканевый обмен (пирацетам, аминалон, ацефен, церебролизин), лечение которыми следует периодически повторять.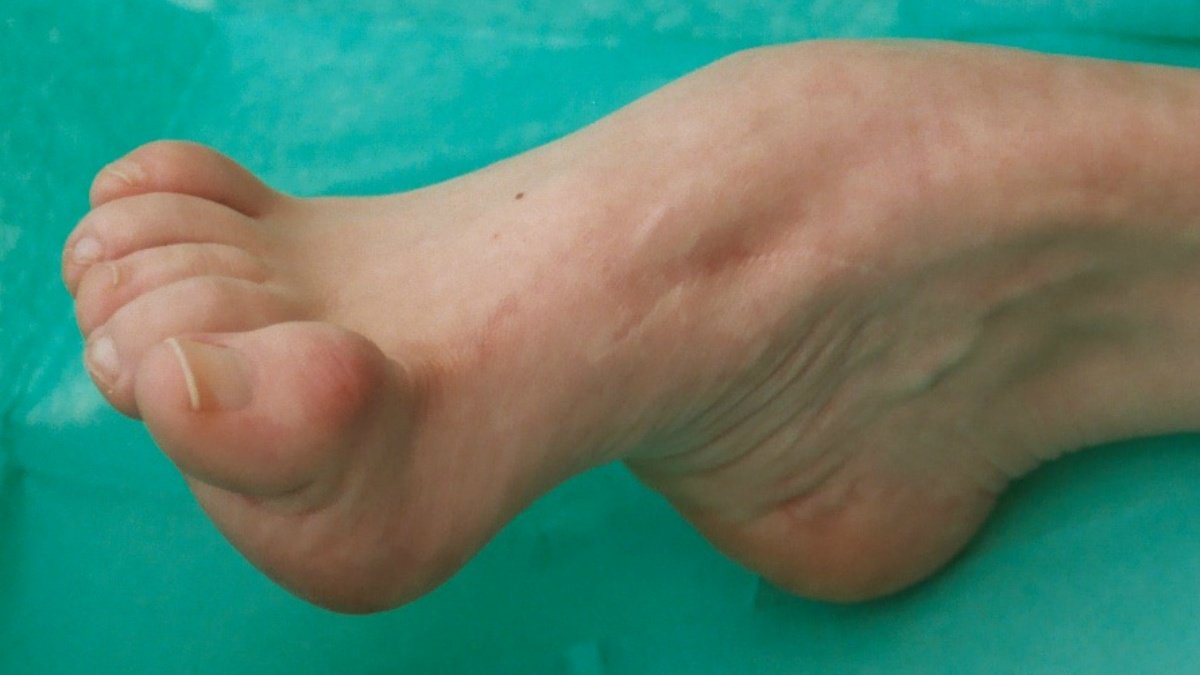
Профилактика атаксии Фридрейха — Особое значение имеет ДНК тестирование на ранней пресимптомной стадии с целью назначения превентивной терапии. Обследуются в первую очередь родственники больного.
Распространенность аутосомно-рецессивной атаксии Фридрейха среди якутского населения PC (Я) составляет 2,8 на 100 тыс. населения. Молекулярно-генетической причиной АФ у якутов является экспансия (GAA) n- повторов в 1 интроне гена FRDA. В улусах вилюйской и центральной Якутии зарегистрированы единичные случаи аутосомно-рецессивной атаксии Фридрейха без накопления в отдельных улусах. Интересен тот факт, что заболевание зарегистрировано у якутского этноса, являющегося представителем азиатской расы, среди которых ранее не было зарегистрировано случаев атаксии Фридрейха.. Это может быть объяснено привнесением в генофонд якутов европейской компоненты, поэтому выяснение причин возникновения и распространения атаксии Фридрейха среди якутов требует дальнейшего изучения.
Литература:
- ДНК-диагностика атаксии Фридрейха с клинико-генетическим анализом.
 // В. В. Пугачев, С. Н. Иллариошкин, Л. В. Прокутша, Б. Д. Маркова, 0. В. Евграфов, И. А. Ишнова-Смоленская // Сборник «Молекулярная диагностика наследственных заболеваний и медико-генетическое консультирование. «- М — MOlIHKll — 1QS3.
// В. В. Пугачев, С. Н. Иллариошкин, Л. В. Прокутша, Б. Д. Маркова, 0. В. Евграфов, И. А. Ишнова-Смоленская // Сборник «Молекулярная диагностика наследственных заболеваний и медико-генетическое консультирование. «- М — MOlIHKll — 1QS3. - Наследственные атаксии и параплегии // С.Н. Иллариошкин. // «Медицина», 2006.
- Наследственные болезни нервной системы.// Ю.Е. Вельтищев, П.А.Темин. // «Медицина», 1998.
- Неврология детского возраста // А.С. Петрухин. // «Медицина», 2004.
- Неврология. Справочник практического врача // Д.Р.Штульман, О.С. Левин // Издательство «МЕДпресс-информ», 2005г.
- Обнаружение полиморфного маркера в области гена, ответственного за возникновение атаксии Фридрейха. // О. В. Евграфов, В. В. Пугачев, Л Б. Стрельченко. // 2-ой Всесоюзный симпозиум «Теоретические и прикладные аспекты молекулярной биологии». Самарканд. — 1991.- тезисы дом. — С. 68.
- Поиск и изучение микросателлитных повторов в области предполагаемой локализации гена FRDA // О. В. Евграфов, В. В. Пугачев, А.В.Поляков, Л Б. Стрельченко // II съезд ВОГИС, Минск, 1992, Тезисы докл. ч.1.с.25
 // В. В. Пугачев, С. Н. Иллариошкин, Л. В. Прокутша, Б. Д. Маркова, 0. В. Евграфов, И. А. Ишнова-Смоленская // Сборник «Молекулярная диагностика наследственных заболеваний и медико-генетическое консультирование. «- М — MOlIHKll — 1QS3.
// В. В. Пугачев, С. Н. Иллариошкин, Л. В. Прокутша, Б. Д. Маркова, 0. В. Евграфов, И. А. Ишнова-Смоленская // Сборник «Молекулярная диагностика наследственных заболеваний и медико-генетическое консультирование. «- М — MOlIHKll — 1QS3. В. Евграфов, В. В. Пугачев, А.В.Поляков, Л Б. Стрельченко // II съезд ВОГИС, Минск, 1992, Тезисы докл. ч.1.с.25
В. Евграфов, В. В. Пугачев, А.В.Поляков, Л Б. Стрельченко // II съезд ВОГИС, Минск, 1992, Тезисы докл. ч.1.с.25Атаксия Фридрейха — причины, симптомы, диагностика и лечение
Атаксия Фридрейха — генетическое заболевание, связанное с нарушением транспорта железа из митохондрий и протекающее с преимущественным поражением клеток центральной и периферической нервной системы, кардиомиоцитов, β — клеток поджелудочной железы, клеток костной ткани и сетчатки. Атаксия Фридрейха диагностируется при помощи МРТ головного и спинного мозга, нейрофизиологических исследований, генетической диагностики. Дополнительно проводится ЭКГ, УЗИ сердца, исследование гормонального фона, рентгенография позвоночника. Лечится атаксия Фридрейха метаболическими и симптоматическими препаратами, соблюдением диеты, регулярными занятиями ЛФК. Хирургическое лечение применяется для устранения костных деформаций.
Общие сведения
Атаксия Фридрейха была описана в 1860 году немецким врачом, имя которого заболевание носит до сих пор.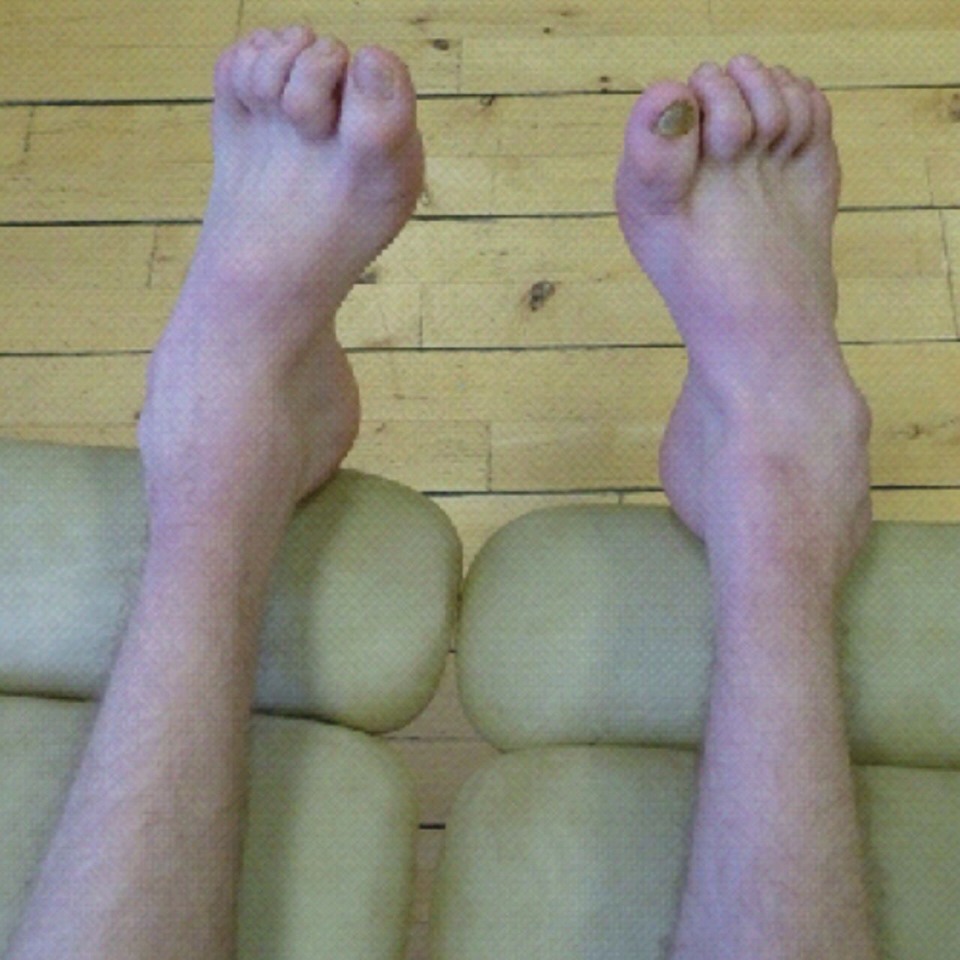 Атаксия Фридрейха относится к группе атаксий, в которую также входят мозжечковая атаксия, атаксия Пьера-Мари, синдром Луи-Бар, корковая и вестибулярная атаксии. В этой группе атаксия Фридрейха является самым распространенным заболеванием. Ее распространенность по всему миру составляет 2-7 заболевших на 100 тыс. населения. У представителей негроидной расы атаксия Фридрейха не отмечается.
Атаксия Фридрейха относится к группе атаксий, в которую также входят мозжечковая атаксия, атаксия Пьера-Мари, синдром Луи-Бар, корковая и вестибулярная атаксии. В этой группе атаксия Фридрейха является самым распространенным заболеванием. Ее распространенность по всему миру составляет 2-7 заболевших на 100 тыс. населения. У представителей негроидной расы атаксия Фридрейха не отмечается.
Атаксия Фридрейха сопровождается поражением не только нервной системы, но и экстраневральными нарушениями. Патологические изменения происходят в сердце, органе зрения, эндокринной системе и опорно-двигательном аппарате. По этой причине атаксия Фридрейха представляет интерес для специалистов в различных областях медицины: неврологии, кардиологии, офтальмологии, эндокринологии, ортопедии и травматологии.
Атаксия Фридрейха
Причины атаксии Фридрейха
Атаксия Фридрейха является генетическим заболеванием и связана с мутацией 9-ой хромосомы, в результате которой отмечается недостаточность или неполноценность белка фратаксина. Этот белок отвечает за транспорт железа из митохондрий. Нарушение его функции приводит к накоплению большого количества железа внутри митохондрий и увеличению свободных радикалов внутри клетки. Последние оказывают повреждающее воздействие на клетку. При этом страдают наиболее активные клетки организма: нейроны (нервные клетки), миокардиоциты (клетки сердечной мышцы), синтезирующие инсулин β — клетки поджелудочной железы, рецепторные клетки сетчатки (палочки и колбочки) и клетки костной ткани. Поражение этих клеток приводит к развитию характерных для атаксии Фридрейха симптомов со стороны периферической и центральной нервной системы, сахарного диабета, кардиомиопатии, нарушений зрения, костных деформаций.
Этот белок отвечает за транспорт железа из митохондрий. Нарушение его функции приводит к накоплению большого количества железа внутри митохондрий и увеличению свободных радикалов внутри клетки. Последние оказывают повреждающее воздействие на клетку. При этом страдают наиболее активные клетки организма: нейроны (нервные клетки), миокардиоциты (клетки сердечной мышцы), синтезирующие инсулин β — клетки поджелудочной железы, рецепторные клетки сетчатки (палочки и колбочки) и клетки костной ткани. Поражение этих клеток приводит к развитию характерных для атаксии Фридрейха симптомов со стороны периферической и центральной нервной системы, сахарного диабета, кардиомиопатии, нарушений зрения, костных деформаций.
Атаксия Фридрейха наследуется аутосомно-рецессивным путем. Носителем обуславливающей ее генной мутации по некоторым данным является 1 человек из 120. Но атаксия Фредрейха развивается лишь в том случае, если человек наследует искаженный ген и от отца, и от матери. При этом его родители являются только носителями генетического нарушения и сами не болеют атаксией Фредрейха.
Симптомы атаксии Фридрейха
Как правило, атаксия Фридрейха начинает проявляться на первых двух десятилетиях жизни. В гораздо более редких случаях наблюдается появление признаков заболевания на третьем или четвертом десятке. Обычно атаксия Фридрейха развивается до 25-летнего возраста. Она начинается с неврологических нарушений и характеризуется неуклонным прогрессированием патологического процесса с усугублением его клинических проявлений.
Атаксия Фридрейха дебютирует нарушениями ходьбы и равновесия. В этот период пациенты отмечают появление шаткости и неуверенности во время ходьбы. Их походка становится неловкой, сопровождается частыми спотыканиями и падениями. Затем возникает нарушение координации при движениях руками, появление тремора рук и связанного с этим изменения почерка. Постепенно присоединяются слабость в ногах, нарушения речи (дизартрия) и снижение слуха (тугоухость). Речь пациентов с атаксией Фридрейха становиться замедленной и невнятной.
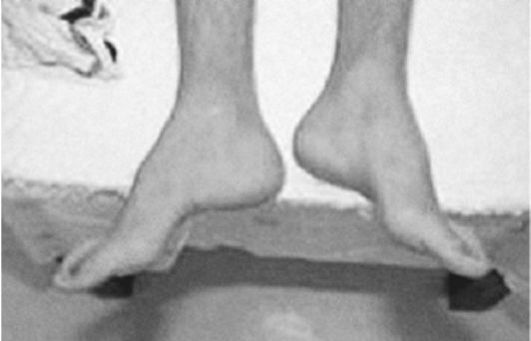
В неврологическом статусе при атаксии Фридрейха отмечается мозжечковый и сенсетивный характер атаксии. Пациент неустойчив в позе Ромберга, не может выполнить пяточно-коленную пробу, промахивается при выполнении пальце-носовой пробы. Результаты проб ухудшаются при их выполнении с закрытыми глазами, поскольку зрение частично компенсирует недостаток координации. Ранним признаком атаксии Фридрейха является исчезновение ахилловых и коленных рефлексов. Характерно наличие симптома Бабинского — разгибания большого пальца стопы при раздражении наружного края подошвы. Иногда разгибание большого пальца сопровождается веерным расхождением остальных пальцев стопы. Симптом Бабинского свидетельствует о поражении пирамидного проводящего пути, отвечающего за двигательную активность.
При прогрессировании атаксии Фридрейха отмечается тотальная арефлексия — отсутствие всех надкостничных и сухожильных рефлексов, расстройство глубоких видов чувствительности (вибрационной чувствительности и суставно-мышечного чувства), снижение мышечного тонуса, слабость (парезы) и атрофические изменения мышц дистальных (расположенных дальше от тела) отделов нижних конечностей. В поздней стадии атаксии Фридрейха парезы, мышечная гипотония и атрофия распространяются на верхние конечности. При этом пациенты утрачивают способность к самообслуживанию. Возможно появление тазовых нарушений и развитие деменции (слабоумия). В ряде случаев атаксия Фридрейха сопровождается снижением слуха, нистагмом, атрофией зрительных нервов.
В поздней стадии атаксии Фридрейха парезы, мышечная гипотония и атрофия распространяются на верхние конечности. При этом пациенты утрачивают способность к самообслуживанию. Возможно появление тазовых нарушений и развитие деменции (слабоумия). В ряде случаев атаксия Фридрейха сопровождается снижением слуха, нистагмом, атрофией зрительных нервов.
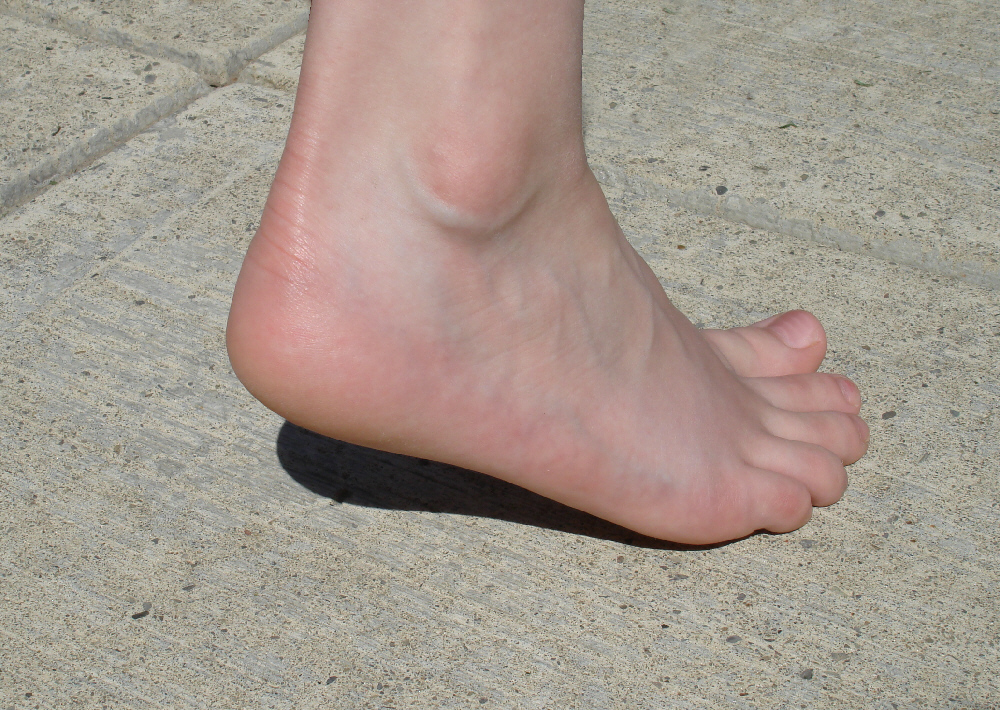
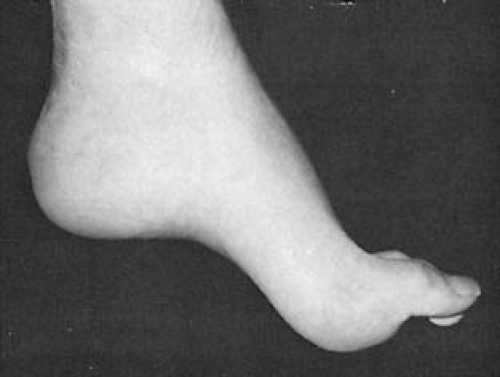
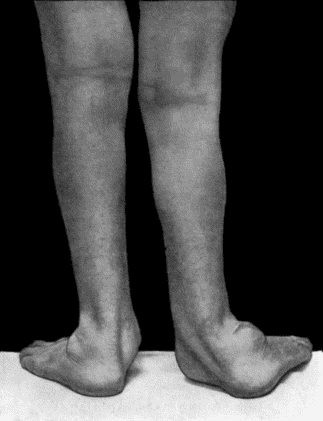
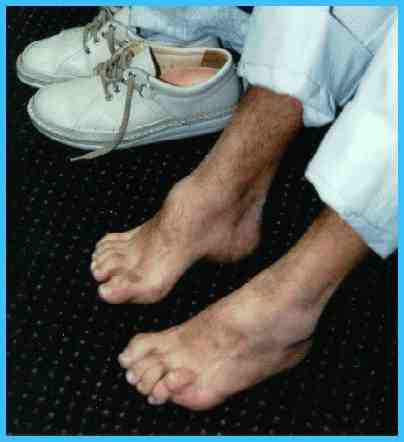
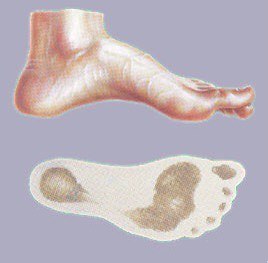
Из экстраневральных клинических симптомов, которыми проявляется атаксия Фридрейха, в 90% случаев наблюдается поражение сердечной мышцы — кардиомиопатия, приводящая к возникновению аритмии (экстрасистолии, пароксизмальной тахикардии, мерцательной аритмии) и сердечной недостаточности. Для атаксии Фридрейха также характерны различные костные деформации. Наиболее типичной является «стопа Фридрейха», имеющая чрезмерно высокий и вогнутый свод, согнутые дистальные фаланги пальцев и разогнутые основные фаланги. Отмечается также сколиоз, косолапость, деформации пальцев рук и ног. Со стороны эндокринной системы атаксия Фридрейха часто сопровождается сахарным диабетом, инфантилизмом, гипогонадизмом, дисфункцией яичников.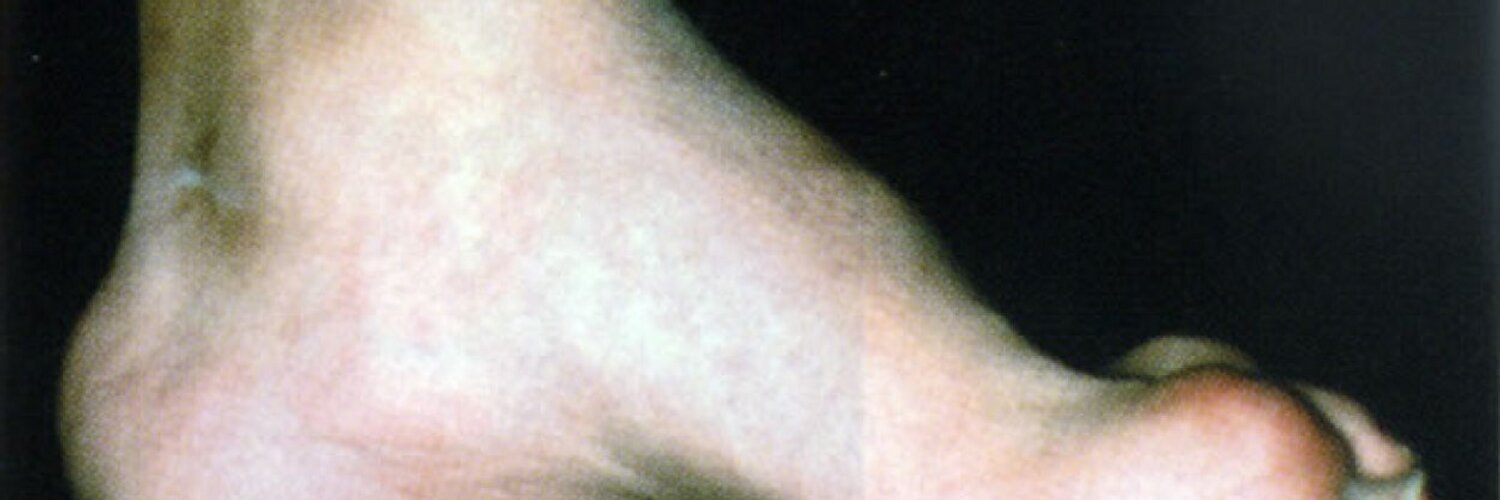 В некоторых случаях у пациентов с атаксией Фридрейха наблюдается катаракта.
В некоторых случаях у пациентов с атаксией Фридрейха наблюдается катаракта.
Диагностика
Диагностика заболевания наиболее затруднительна в случаях, когда атаксия Фридрейха начинается с экстраневральных проявлений. При этом некоторые пациенты в течении нескольких лет наблюдаются у кардиолога по поводу заболевания сердца или у ортопеда по поводу сколиоза. Лишь при развитии неврологической симптоматики они попадают на консультацию к неврологу.
Основными методами инструментальной диагностики атаксии Фридрейха является магнитно-резонансная томография и нейрофизиологическое тестирование. МРТ головного мозга выявляет атрофические процессы в продолговатом мозге и мосте, атрофию мозжечка. При МРТ позвоночника наблюдается уменьшение поперечника спинного мозга и его атрофические изменения. В постановке диагноза «атаксия Фридрейха» КТ головного мозга не достаточно информативна. С ее помощью характерные изменения можно визуализировать только в поздних стадиях заболевания. Ранняя атаксия Фридрейха сопровождается только КТ-признаками незначительной атрофии мозжечка.
Ранняя атаксия Фридрейха сопровождается только КТ-признаками незначительной атрофии мозжечка.
Исследование проводящих путей проводится при помощи транскраниальной магнитной стимуляции, исследование периферических нервов — путем электронейрографии и электромиографии. При этом атаксия Фридрейха характеризуется умеренным снижением потенциалов действия при проведении по двигательным нервам в сочетании с большим (вплоть до полного исчезновения) снижением проводимости по чувствительным волокнам.
По причине наличия экстраневральных проявлений атаксия Фредрейха требует дополнительных исследований сердечно-сосудистой, эндокринной и опорно-двигательной систем. С этой целью проводится консультация кардиолога, ортопеда, офтальмолога и эндокринолога; анализ сахара крови и глкозо-толерантный тест, исследование гормонального фона; ЭКГ, нагрузочные тесты, УЗИ сердца; рентгенография позвоночника.
Немаловажное значение в установлении диагноза «атаксия Фридрейха» имеет медико-генетическое консультирование и комплексная (прямая и косвенная) ДНК-диагностика. Она производится на образцах крови пациента, его биологических родителей, а также кровных братьев и сестер. В период ведения беременности атаксия Фридрейха у плода может быть диагностирована при помощи ДНК-диагностики ворсин хориона на 8-12 неделе беременности или амниотической жидкости на 16-24 неделе.
Она производится на образцах крови пациента, его биологических родителей, а также кровных братьев и сестер. В период ведения беременности атаксия Фридрейха у плода может быть диагностирована при помощи ДНК-диагностики ворсин хориона на 8-12 неделе беременности или амниотической жидкости на 16-24 неделе.
Атаксия Фридрейха требует дифференциальной диагностики с фуникулярным миелозом, опухолью мозжечка, нейросифилисом, обменными наследственными заболеваниями (болезнью Ниманна-Пика, болезнью Краббе, синдромом Луи-Бар, наследственным дефицитом витамина Е), рассеянным склерозом.
Лечение атаксии Фридрейха
Адекватное и регулярной лечение атаксии Фридрейха позволяет приостановить прогрессирование заболевания, избежать осложнений, длительное время сохранять способность пациента вести активный образ жизни. Как правило, атаксия Фридрейха лечится одновременным назначением метаболических препаратов, принадлежащих к 3 различным группам: кофакторов энергетических энзимных реакций, стимуляторов активности дыхательной цепи митохондрий и антиоксидантов.
Дополнительно при атаксии Фридрейха назначаются медикаменты, улучшающие метаболические процессы в сердечной мышце (тиаминпирофосфат, инозин, триметазидин, 5-гидроксипрофан и пр), ноотропы и нейропротекторы (гамма-аминомасляная кислота, пирацетам, меклофеноксат, пиритинол), поливитамины. При необходимости в пораженные мышцы вводится ботулотоксин, осуществляются хирургические операции по коррекции костных деформаций.
Большое значение для пациентов с атаксией Фридрейха имеет лечебная физкультура. Постоянные занятия ЛФК, направленные на тренировку координации и мышечной силы, дают возможность сохранять двигательную активность и купировать возникающие болезненные ощущения. Поскольку атаксия Фридрейха сопровождается нарушением энергетического обмена, то пациентам с этим заболеванием необходимо ограничить прием углеводов с пищей, избыток которых может провоцировать усугубление обменных нарушений.
Прогноз атаксии Фридрейха
Атаксия Фридрейха имеет неуклонно прогрессирующее течение, приводящее к летальному исходу. Пациент погибает от сердечной или дыхательной недостаточности, инфекционных осложнений. Около 50% пациентов, у которых наблюдается атаксия Фридрейха, не доживают до 35-летнего возраста. У женщин течение заболевания более благоприятно. Продолжительность их жизни в 100% составляет более 20 лет от момента начала атаксии, в то время как среди мужчин лишь 63% живут дольше этого срока, В крайне редких случаях при отсутствии сердечных нарушений и сахарного диабета пациенты живут до 70-80 лет.
Пациент погибает от сердечной или дыхательной недостаточности, инфекционных осложнений. Около 50% пациентов, у которых наблюдается атаксия Фридрейха, не доживают до 35-летнего возраста. У женщин течение заболевания более благоприятно. Продолжительность их жизни в 100% составляет более 20 лет от момента начала атаксии, в то время как среди мужчин лишь 63% живут дольше этого срока, В крайне редких случаях при отсутствии сердечных нарушений и сахарного диабета пациенты живут до 70-80 лет.
Атаксия Фридрейха — описание болезни наследственная атаксия, признаки и причины заболевания ataxia friedreich, а также профилактика при осложнении и определение рецидива, диагноз и симптомы болезни неустойчивость походки
Заболевание начинается в возрасте 8-15 лет с шаткости при хотьбе и частых падений. Атаксия Фридрейха составляет половину всех заболеваний этой группы. Половина больных не доживает до 35 лет. Прогноз благоприятнее у женщин: более 20 лет с начала заболевания живут 100% женщин и только 63% мужчин.
Заболевание начинается до 25 лет и характеризуется неустойчивостью при хотьбе, частыми падениями и тремором туловища, нистагмом. Неврологические нарушения больше выражены в ногах. Иногда первым признаком заболевания может быть дизартрия, атаксия, мышечная гипотония, мышечная дистония, нарушения глубокой чувствительности, сухожильная арефлексия, амиотрофии, парезы, электрофизиологические признаки сенсорной полиневропатии. Поражение сердца (кардиомегалия , симметричная гипертрофия миокарда , шум и нарушение проводимости) наблюдается у 90% больных. Изредка наблюдаются умственная отсталость или иные психические расстройства . У 20% больных обнаруживают сахарный диабет. Характерны деформации скелета : полая стопа , косолапость и сколиоз.
Симптомы: неустойчивость походки, тремор, неконтролируемые движения глаз (нистагм), атаксия (отсутствие координации), мышечная гипотония, снижение чувствительности, парезы ног, кардиомегалия, шумы в сердце, изменение психического состояния, сахарный диабет, деформация стопы, искривление позвоночника, деформация костей
Дагноз может быть заподозрен клинически. Проводят медико-генетическое консультирование, ДНК-диагностику, МРТ, рентгенографию позвоночника, конечностей, ЭКГ, ЭЭГ, электронейромиографию, определение толерантности к глюкозе, цитохимичекое исследование ( активность фермента сукцинатдегидрогеназы СДГ)
Проводят медико-генетическое консультирование, ДНК-диагностику, МРТ, рентгенографию позвоночника, конечностей, ЭКГ, ЭЭГ, электронейромиографию, определение толерантности к глюкозе, цитохимичекое исследование ( активность фермента сукцинатдегидрогеназы СДГ)
Следует различать: атаксия фридрейха, рассеянный склероз, нейросифилис, опухоли головного мозга, болезнь спинного мозга неуточненная
Медицинские процедуры, проводимые при заболевании атаксия фридрейха: Медико-генетическое консультирование, ДНК-тест, Магнитно-резонансная томография, Рентген, ЭКГ, Электроэнцефалограмма, Электромиография, Кровь на сахар, Определение толерантности к глюкозе, Биохимический анализ крови
Прогноз неблагоприятный, средняя продолжительность жизни с начала прогрессирования заболевания 15—20 лет. Очень редко больные доживают до 70—80 лет, при условии отсутствия сопутствующих заболеваний.
Осложнения: кардиомиопатия, легочная (дыхательная) недостаточность, катаракта, деменция, истощение и атрофия мышц, не классифицированные в других рубриках
Лечение атаксии Фридрейха не приводит к полному выздоровлению, но профилактика дает возможность избежать развития многих симптомов и осложнений. Назначают препараты, улучшающие метаболизм в структурах нервной системы (антиоксиданты, общеукрепляющие средства). В целом лечение симптоматическое, направлено на лечение сахарного диабета, заболеваний сердечно-сосудистой системы. Показана физиотерапия и лечебная физкультура. При выраженной деформации стоп показана ортопедическая обувь.
Профилактика — Особое значение имеет ДНК тестирование на ранней досимптомной стадии заболевания с целью назначения превентивной терапии. Обследуются в первую очередь родственники больного.
Спастическая параплегия Штрюмпеля 4.
 73.18.3 GJC2 м.
73.18.3 GJC2 м.Исследуемый материал Цельная кровь (с ЭДТА)
Метод определенияСеквенирование
Выдаётся заключение врача-генетика!
Исследование мутаций в гене GJC2./37.jpg)
Болезнь Штрюмпеля относится к гетерогенным заболеваниям. Описано аутосомно-доминантное, аутосомно-рецессивное и Х-сцепленное наследование заболевания. В большинстве семей имеет место аутосомно-доминантное наследование болезни, соответствующее оригинальным описаниям Штрюмпеля. Именно для аутосомно-доминантной наследственной спастической параплегии обоснованно использование широко распространенного эпонимического термина болезнь Штрюмпеля. Наследственная спастическая параплегия характеризуется глиозным перерождением пирамидных путей в боковых и передних канатиках на грудном, поясничном уровне спинного мозга.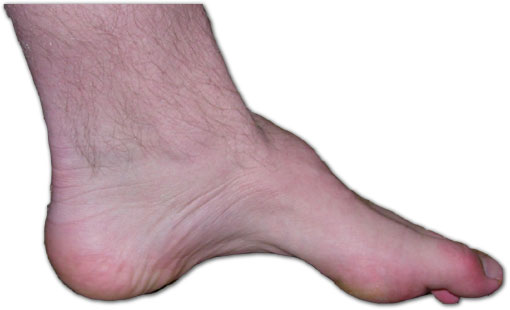 На поздних стадиях болезни вовлекаются пирамидные волокна в стволе мозга и частичная гибель клеток Беца двигательной зоны коры полушарий большого мозга. Первые симптомы могут развиваться в любом возрасте от 1-го до 7-го десятилетия жизни, что связано с генетической гетерогенностью.
На поздних стадиях болезни вовлекаются пирамидные волокна в стволе мозга и частичная гибель клеток Беца двигательной зоны коры полушарий большого мозга. Первые симптомы могут развиваться в любом возрасте от 1-го до 7-го десятилетия жизни, что связано с генетической гетерогенностью.
Тип наследования.
Аутосомно- рецессивный.
Гены, ответственные за развитие заболевания.
Ген GJC2 белка щелевых контактов коннексин 47 (Сх47) (GAP JUNCTION PROTEIN, GAMMA-2). Ген расположен на хромосоме 1 в регионе 1q42.13. Содержит 2 экзона.
Мутации в этом гене приводят также к развитию гипомиелинизирующей лейкодистрофии тип 2, врождённой лимфедеме, тип 1С.
Патогенез и клиническая картина.
Наследственная спастическая параплегия характеризуется глиозным перерождением пирамидных путей в боковых и передних канатиках на грудном, поясничном уровне спинного мозга, на поздних стадиях болезни вовлекаются пирамидные волокна в стволе мозга и частичная гибель клеток Беца двигательной зоны коры полушарий большого мозга.
Первые симптомы могут развиваться в любом возрасте от 1-го до 7-го десятилетия жизни, что связано с генетической гетерогенностью.
Для болезни Штрюмпеля характерны следующие признаки проявления заболевания: постепенное развитие заболевания, скованность, быстрая утомляемость ног при ходьбе, стягивающие судороги в мышцах ног, спастическая походка, повышение сухожильных рефлексов, клонусы стоп и коленных чашечек, раннее появление сухожильных рефлексов, формирование контрактур и деформация стопы по типу «стопы Фридрейха». Верхние конечности вовлекаются редко и в поздней стадии. Чувствительность и интеллект не изменены. Иногда – поражение II (зрительный) и III (глазодвигательный) нервов, дизартрия, нистагм, атаксия, интенционный тремор.
Верхние конечности вовлекаются редко и в поздней стадии. Чувствительность и интеллект не изменены. Иногда – поражение II (зрительный) и III (глазодвигательный) нервов, дизартрия, нистагм, атаксия, интенционный тремор.
Характерным параклиническим признаком наследственной спастической параплегии является картина атрофии спинного мозга на всем протяжении (особенно в каудальных отделах), выявляемая при проведении МРТ.
Для данного типа заболевания характерно: позднее начало заболевания, медленно прогрессирующее течением, осложненное спастической параплегией, с нормальным или почти нормальным психомоторным развитием, нистагм отсутствует. Способность ходить больные сохраняют до зрелого возраста. Гетерозиготные члены семьи здоровы.
Частота встречаемости: заболевание редкое. Описана одна семья.
Перечень исследуемых мутаций может быть предоставлен по запросу.
Литература
- GE Rudenskaya, AV Polyakov. Hereditary spastic paraplegia in Russia: epidemiological, clinical and molecular aspects. TWS symposium on hereditary spastic paraplegias: www.hsp-info.de.
- Orthmann-Murphy, J. L., Salsano, E., Abrams, C. K., Bizzi, A., Uziel, G., Freidin, M. M., Lamantea, E., Zeviani, M., Scherer, S. S., Pareyson, D. Hereditary spastic paraplegia is a novel phenotype for GJA12/GJC2 mutations. Brain 132: 426-438, 2009.
- OMIM.
 Hereditary spastic paraplegia in Russia: epidemiological, clinical and molecular aspects. TWS symposium on hereditary spastic paraplegias: www.hsp-info.de.
Hereditary spastic paraplegia in Russia: epidemiological, clinical and molecular aspects. TWS symposium on hereditary spastic paraplegias: www.hsp-info.de.причины, симптомы, диагностика, лечение, профилактика
Представляет собой генетическое заболевание, обусловленное нарушением транспорта железа из митохондрий и характеризующееся преимущественным поражением клеток центральной и периферической нервных систем, клеток поджелудочной железы, костной ткани и сетчатки глаза.
ПричиныАтаксия Фридрейха представляет собой наследственное заболевание, возникающее на фоне мутации 9-ой хромосомы, в результате чего развивается недостаточность либо неполноценность фратаксина, белка, который корректирует вывод железа из митохондрий. Расстройство функции этого белка сопровождается накоплением значительного количества железа внутри митохондрий, а также повышения внутри клетки концентрации свободных радикалов, которые оказывают повреждающее воздействие на клетку. В следствии таких процессов страдают самые активные клетки организма, такие как нейроны, миокардиоциты, β-клетки поджелудочной железы, продуцирующие инсулин, рецепторные клетки сетчатки и клетки костной ткани. В результате поражения этих клеток отмечается развитие характерных для атаксии Фридрейха симптомов со стороны периферической и центральной нервной системы, а также сахарного диабета, кардиомиопатии, нарушения зрения и костных деформаций.
Расстройство функции этого белка сопровождается накоплением значительного количества железа внутри митохондрий, а также повышения внутри клетки концентрации свободных радикалов, которые оказывают повреждающее воздействие на клетку. В следствии таких процессов страдают самые активные клетки организма, такие как нейроны, миокардиоциты, β-клетки поджелудочной железы, продуцирующие инсулин, рецепторные клетки сетчатки и клетки костной ткани. В результате поражения этих клеток отмечается развитие характерных для атаксии Фридрейха симптомов со стороны периферической и центральной нервной системы, а также сахарного диабета, кардиомиопатии, нарушения зрения и костных деформаций.
Развитие атаксия Фредрейха возможно только в том случае, если ребенок получает мутированный ген от обеих родителей. В этом случае родители являются только носителями патологического гона, но при этом сами не страдают данным недугом.
СимптомыРазвитие атаксия Фредрейха отмечается в первые двадцать лет жизни ребенка.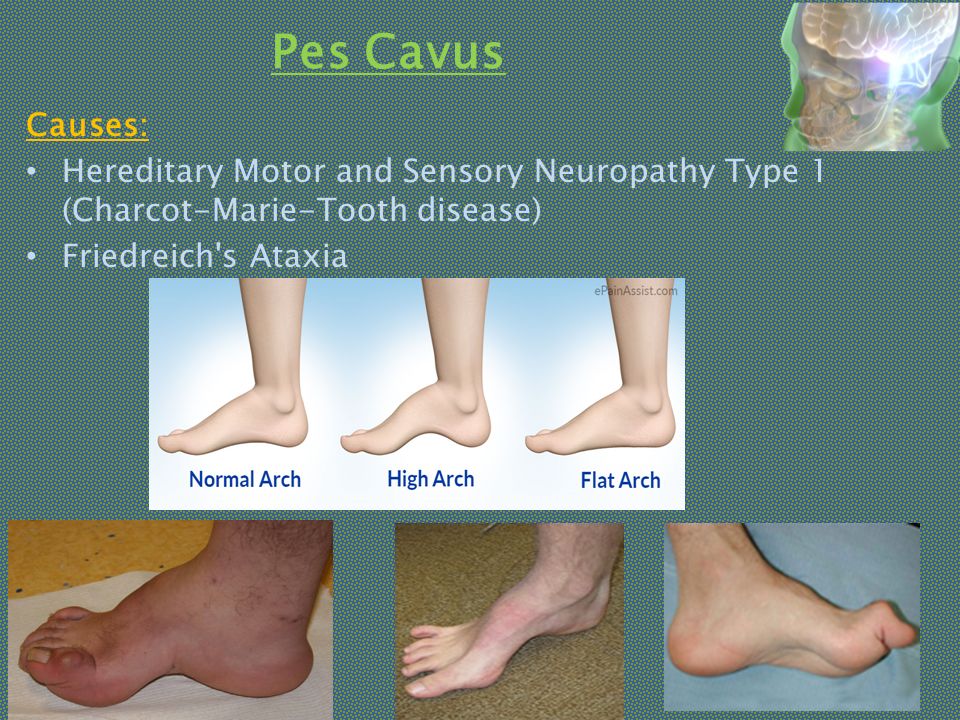 В более редких случаях манифестация заболевания может возникать на третьем либо четвертом десятке жизни человека. Чаще всего дебют атаксии происходит до 25 лет с возникновения прогрессирующих неврологических нарушений.
В более редких случаях манифестация заболевания может возникать на третьем либо четвертом десятке жизни человека. Чаще всего дебют атаксии происходит до 25 лет с возникновения прогрессирующих неврологических нарушений.
У таких больных отмечается нарушение походки и равновесия. На этом этапе у больного отмечается появление шаткости при ходьбе, а также частых спотыканий и падений. По мере прогрессирования заболевания отмечается появление нарушения координации при движении руками, развитие тремора и выраженной слабости в ногах, а также нарушения речи, ухудшения слуха, у них отмечается появление невнятности и замедления речи.
При определении неврологического статуса при этом заболевании отмечается возникновение мозжечковой и сенсетивной атаксии. У таких больных отмечается появление неустойчивости в позе Ромберга, они не способны выполнить пяточно-коленную пробу, а пальценосовую пробу выполняют с мимопромахиванием, при этом отмечается значительное ухудшение результатов пробы при выполнении ее с закрытыми глазами./10be22d109d172f.s.siteapi.org/img/8cb9cd9685639e3dbec021cc025324f9a444935f.JPG) Одним из ранних симптомов атаксии Фридрейха считается исчезновение ахилловых и коленных рефлексов. У таких больных характерно наличие симптома Бабинского, обусловленного разгибанием большого пальца стопы при раздражении наружного края подошвы. В редких случаях разгибание большого пальца стопы может сопровождаться веерным расхождением остальных пальцев стопы. Симптом Бабинского указывает на поражении пирамидного проводящего пути, который отвечает за двигательную активность.
Одним из ранних симптомов атаксии Фридрейха считается исчезновение ахилловых и коленных рефлексов. У таких больных характерно наличие симптома Бабинского, обусловленного разгибанием большого пальца стопы при раздражении наружного края подошвы. В редких случаях разгибание большого пальца стопы может сопровождаться веерным расхождением остальных пальцев стопы. Симптом Бабинского указывает на поражении пирамидного проводящего пути, который отвечает за двигательную активность.
Тяжелая форма заболевания обусловлена развитием тотальной арефлексии, снижения мышечного тонуса, слабости и атрофических изменений в мышца дистальных отделов нижних конечностей. На поздних стадиях атаксии Фридрейха у больного формируются парезы, мышечная гипотония, а атрофия мышц может распространяться на верхние конечности. Больные утрачивают способность к самообслуживанию, у них может отмечаться появление тазовых нарушений и развитие деменции.
ДиагностикаДиагностика атаксии Фридрейха основана на назначении больному проведения магнитнорезонансной томографии и нейрофизиологического тестирования. При проведении магниторезонансной томографии в головном мозге таких больных происходит выявление атрофических процессов в продолговатом мозге и мосте, а также атрофия мозжечка. При обследовании позвоночника отмечается уменьшение поперечника спинного мозга и его атрофические изменения. Также таким больным может потребоваться проведение исследования проводящих путей посредством транскраниальной магнитной стимуляции, исследования периферических нервов при помощи электронейрографии и электромиографии.
При проведении магниторезонансной томографии в головном мозге таких больных происходит выявление атрофических процессов в продолговатом мозге и мосте, а также атрофия мозжечка. При обследовании позвоночника отмечается уменьшение поперечника спинного мозга и его атрофические изменения. Также таким больным может потребоваться проведение исследования проводящих путей посредством транскраниальной магнитной стимуляции, исследования периферических нервов при помощи электронейрографии и электромиографии.
Назначение корректного лечение атаксии Фридрейха позволяет приостановить прогрессирование заболевания, избежать осложнений, а также на протяжении длительного времени сохранять способность пациента вести активный образ жизни. Лечение заболевания происходит на основе использования метаболических препаратов, принадлежащих к трем различным группам: кофакторов энергетических энзимных реакций, стимуляторов активности дыхательной цепи митохондрий и антиоксидантов.
Также потребуется назначение лекарственных средств, улучшающих метаболические процессы в сердечной мышце, ноотропов, нейропротекторов и поливитаминов.
ПрофилактикаПредупредить развитие у ребенка атаксии Фридрейха поможет посещение генетика при планировании беременности.
Атаксия Фридрейха — причины и признаки атаксии Фридрейха
Атаксия Фридрейха — это генетически обусловленное заболевание, которое появляется в случае неправильной транспортировки железа из митохондрий.
Содержание статьи
Причины
Болезнь имеет наследственный характер.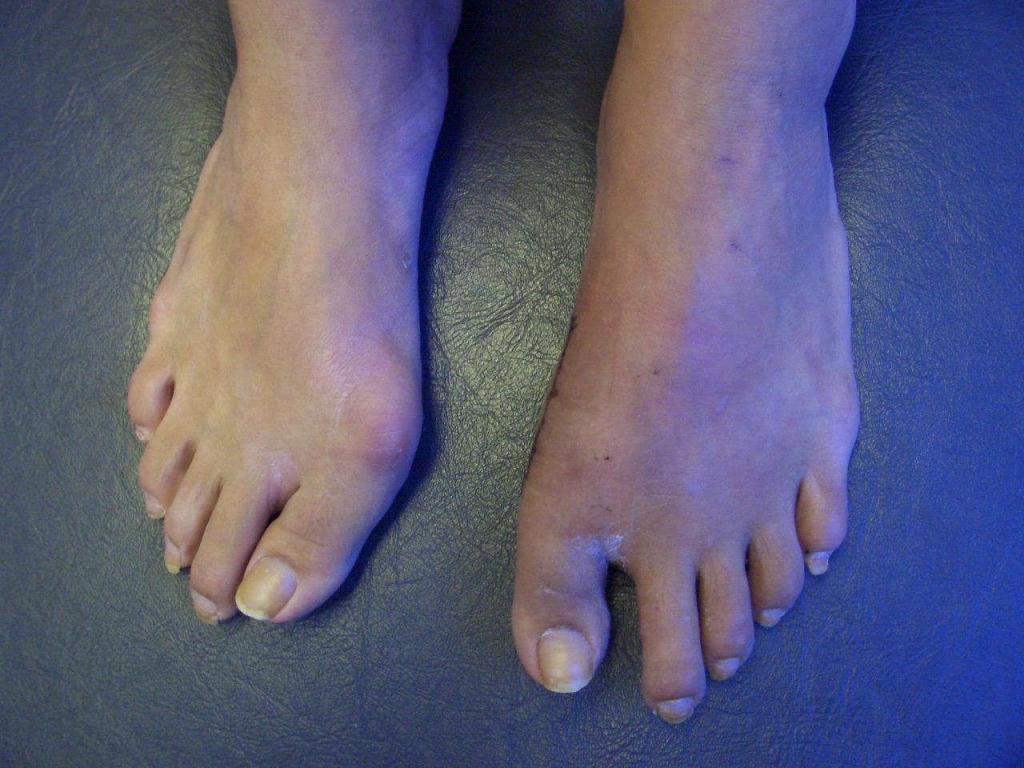 Развитие ее происходит при получении ребенком мутационного гена от обоих родителей, которые являются носителями патологии.
Развитие ее происходит при получении ребенком мутационного гена от обоих родителей, которые являются носителями патологии.
Симптомы
Обычно развитие патологии происходит до 25 лет. Намного реже — в возрасте от тридцати до пятидесяти. Первым признаком является нарушенная ходьба и неспособность удерживать тело в равновесии.
Появляется шаткость и неуверенность при ходьбе. Человек спотыкается и падает. Нарушается координация в процессе движения верхними конечностями, отмечается их тремор, изменяется почерк. К вышеупомянутым признакам присоединяются слуховые и речевые нарушения, чувство слабости в ногах.
Затруднено выполнение пяточно-коленного теста, выражена неустойчивость в позе Ромберга. Человек промахивается в ходе пальце-носового теста. Полностью исчезают ахилловые и коленные рефлексы. Возникает симптом Бабинского — в ходе раздражения наружной части подошвы ступни разгибается большой палец.
В ходе прогрессирования недуга возникает тотальная арефлексия.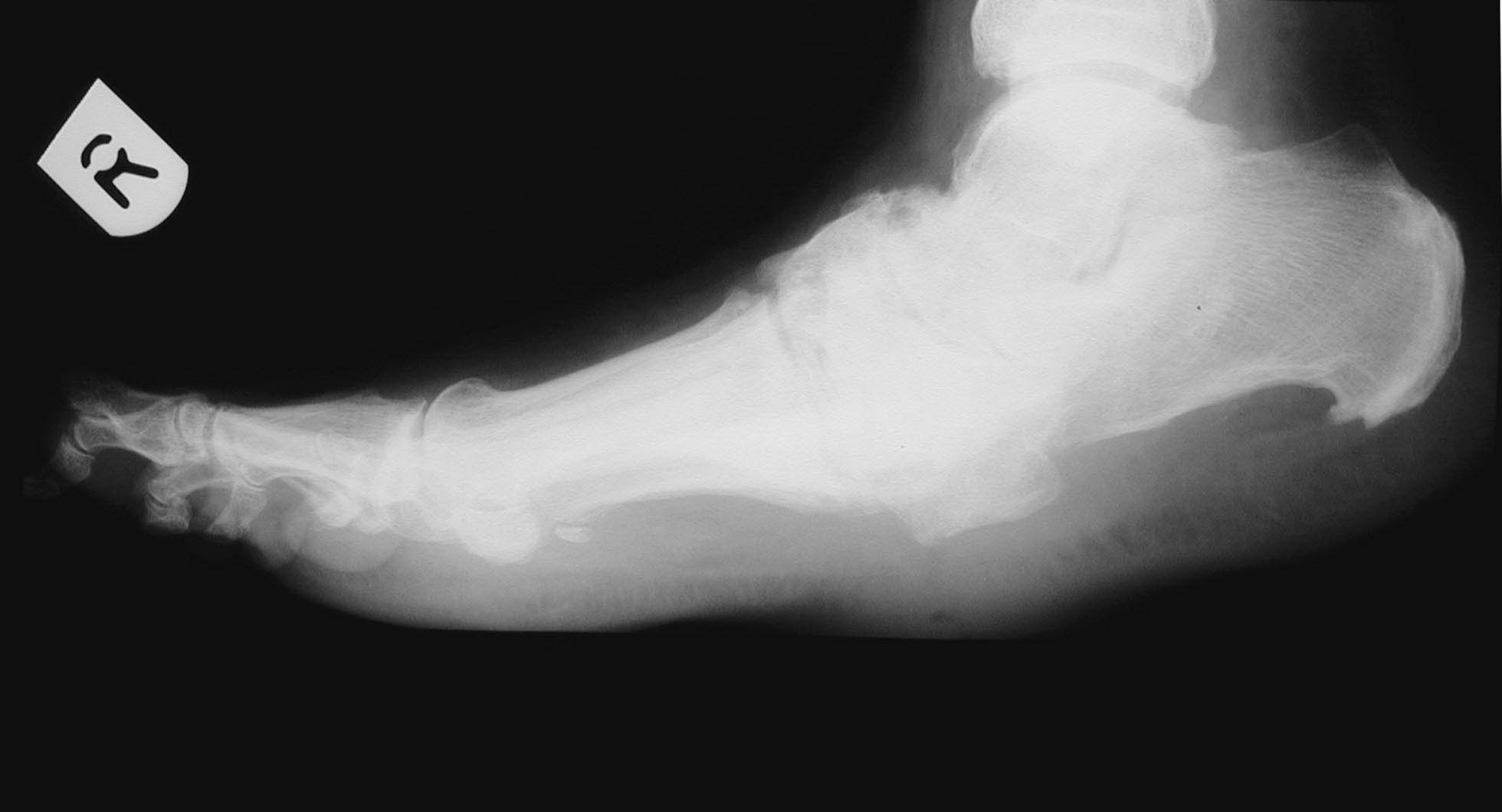 На позднем этапе наблюдается поражение мышц рук атрофией, парезом и гипотонией. Появляются тазовые нарушения. У человека может развиться слабоумие. Иногда отмечается снижение слуха, нистагм, атрофируются зрительные нервы.
На позднем этапе наблюдается поражение мышц рук атрофией, парезом и гипотонией. Появляются тазовые нарушения. У человека может развиться слабоумие. Иногда отмечается снижение слуха, нистагм, атрофируются зрительные нервы.
Для экстраневральной клинической симптоматики характерна кардиомиопатия, которая приводит к аритмии и развитию сердечной недостаточности. Характерны деформации костей: возникновение сколиоза, «стопы Фридрейха», косолапости и пальцевой деформации всех конечностей. Эндокринные нарушения проявляются диабетом, гипогонадизмом, инфантилизмом и дисфункцией яичников. Иногда отмечается появление катаракты.
Диагностика
Диагностика болезни затруднена, если она начинается с экстраневральной симптоматики. Некоторые больные несколько лет могут стоять на учете у кардиолога с сердечными патологиями или у ортопеда со сколиозом. Только при появлении неврологических симптомов следует записаться на прием к неврологу.
Проводится МРТ позвоночника, магнитно-резонансная или компьютерная томография мозга. Назначается нейрофизиологическое тестирование.
Назначается нейрофизиологическое тестирование.
Транскраниальная магнитная стимуляция позволяет исследовать проводящие пути, а электронейрография и электромиографии — периферические нервы .
При необходимости больной обязательно консультируется у эндокринолога, кардиолога, ортопеда или офтальмолога. Берется анализ крови на определение сахара, проводятся глюкозотолерантный тест, рентген позвоночного столба, электрокардиограмма, нагрузочные пробы, УЗИ сердца, исследуется гормональная система.
Для постановки точного диагноза проводится комплексная диагностика ДНК, включающая исследование крови родителей, сестер и братьев, медико-генетическое консультирование. Благодаря ДНК-диагностики также можно выявить патологию у плода.
Лечение
Назначается прием метаболических лекарственных препаратов.
Дополнительно могут выписываться средства, которые способствуют улучшению метаболизма в мышце сердца, а также поливитамины, нейропротекторы и ноотропы и поливитамины. При показании в места поражения вводится ботулотоксин или проводится операция с целью коррекции возникшей деформации костей.
При показании в места поражения вводится ботулотоксин или проводится операция с целью коррекции возникшей деформации костей.
Больному рекомендуется постоянно заниматься ЛФК, что помогает остановить боль и сохранять двигательную активность длительное время.
Из-за того, что течение патологии сопровождает нарушение энергетического обмена, назначается диета с низким содержанием углеводов.
Профилактика
Специфической профилактики для предупреждения заболевания не существует, так как оно передается наследственным путем. Будущим родителям перед планированием беременности рекомендуется сдать тесты на выявление у них мутационного гена.
Невральная амиотрофия Шарко-Мари-Тута
Больше информации про другие виды заболеваний на букву «Н»:
Нарушение сна;
Нарколепсия;
Наследственная мозжечковая атаксия Пьера-Мари;
Нарушения спинномозгового кровообращения;
Невралгия тройничного нерва;
Невралгия подчелюстного и подъязычного узлов;
Невралгия языкоглоточного узла;
Невралгия ушного узла;
Неврастения;
Невральная амиотрофия Шарко-Мари-Тута;
Невринома слухового нерва;
Невринома;
Неврит зрительного нерва;
Неврит глотки;
Неврит лицевого нерва;
Неврит;
Невроз навязчивых состояний;
Невроз глотки;
Неврозы;
Неврозоподобное заикание;
Невропатия бедренного нерва.
Общее понятие
Невральная амиотрофия Шарко-Мари-Тута — хроническое заболевание организма, носящее наследственный тип образования. Характерным показателем является поражение нервной системы периферического отдела. Аномальные процессы проявляются в формировании и изменений структуры в мышечных зонах, например: уменьшение в размерах районов в начале нижних конечностей, а после — верхних. В комбинации с аномалией данного вида у больного может возникнуть гипестезия и снижение рефлекторной способности сухожилий, подергивания разных отделов мышц.
Врачи прибегают к различным вариантам обследования пациента с таким недугом, к ним относятся: электромиография, электронейрография, генетическое обследование, ДНК-проверка, биопсия нервов и мышечных участков. Радикальных приемов лечения — не существует, но имеются методики направленные на избавление человека от симптомов. Медики предписывают разнообразные витаминные комплексы, антихолинэстеразной способы, метаболические приемы, микроциркуляторные аналоги, ЛФК, физиотерапию и прочее.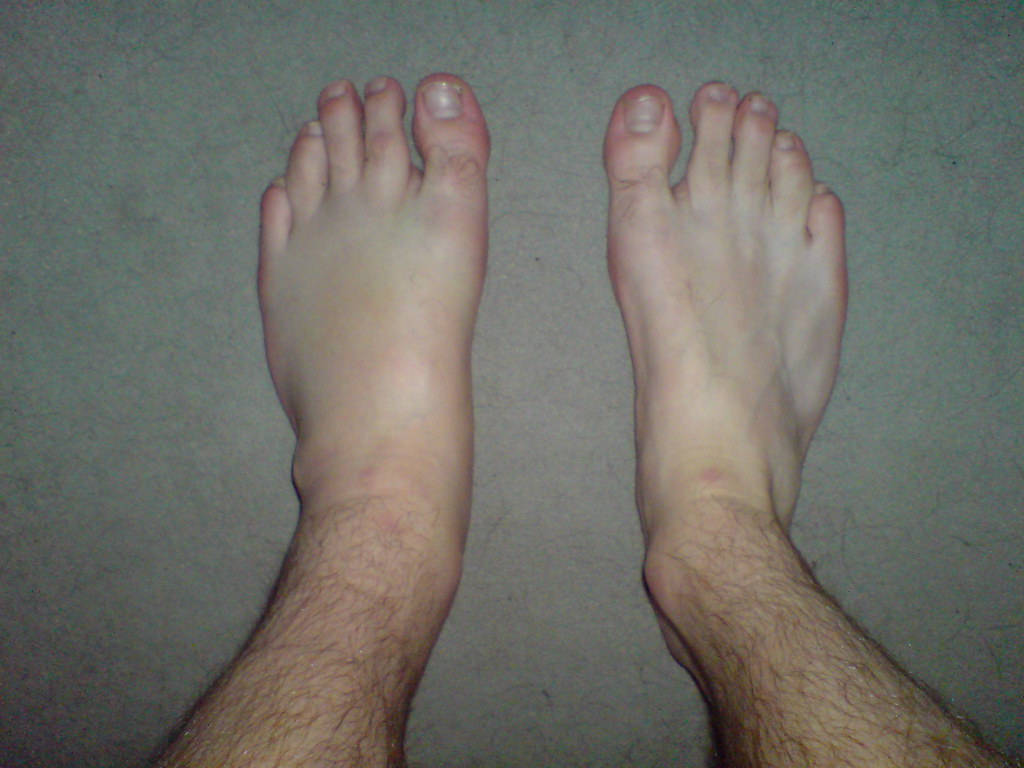
Полезные сведения
Невральная амиотрофия Шарко-Мари-Тута (ШМТ) входит в список хронических заболеваний (полиневропатий) генетического характера, которые проявляются в повышенном прогрессировании. К ней относятся:
- Синдром Русси-Леви.
- Гипертрофическая невропатия Дежерина-Сотта.
- Болезнь Рефсума и другие более редкие патологические процессы.
Согласно среднестатистическим данным ученые заявляют, что практически 83% случая вызваны в результате наследственной предрасположенности человека. Неблагоприятные действия в теле возникают по большей мере у мужчин, женщины менее подвержены описываемому заболеванию. Ссылаясь на многочисленные сведения, невральная амиотрофия ШМТ диагностируется с частотой от двух до тридцати шести случаев на сто тысяч населения планеты. Обычно, она носит семейный характер. Однако у членов одной семьи симптоматика клиника может быть по-разному выражена. Вместе с этим врачи наблюдают и спорадические варианты ШМТ. Медицинские сотрудники отмечают четкую взаимосвязь с потеряй координации по Фридрейху. У некоторых больных в разных ситуациях замечаются типичные симптомы одной или другой болезни. Иногда, с течением долгих лет, клиника одной аномалии может замениться симптомами другой.
У некоторых больных в разных ситуациях замечаются типичные симптомы одной или другой болезни. Иногда, с течением долгих лет, клиника одной аномалии может замениться симптомами другой.
Патогенез невральной амиотрофии Шарко-Мари-Тута
В медицине на данный момент не имеется точной и достоверной информации о происхождении и механизме возникновения невральной амиотрофии. Согласно проведенным экспериментам стало понятно только то, что почти у 75% пациентов с таким диагнозом, которые прошли генетическую консультацию, выделялось повторение определенной зоны 17-й хромосомы. Уже известно, что заболевание обладает несколькими группами, вызываемыми различными мутациями генов. Так, например, во время исследования больного с ШМТ, проявляющейся из-за мутации белка гена MFN2, начинается появление частиц митохондрий. В результате чего у человека происходит сбой в движении этих элементов по аксону.
Большое количество форм проявляются вследствие поражения миелинового наружного слоя волокон.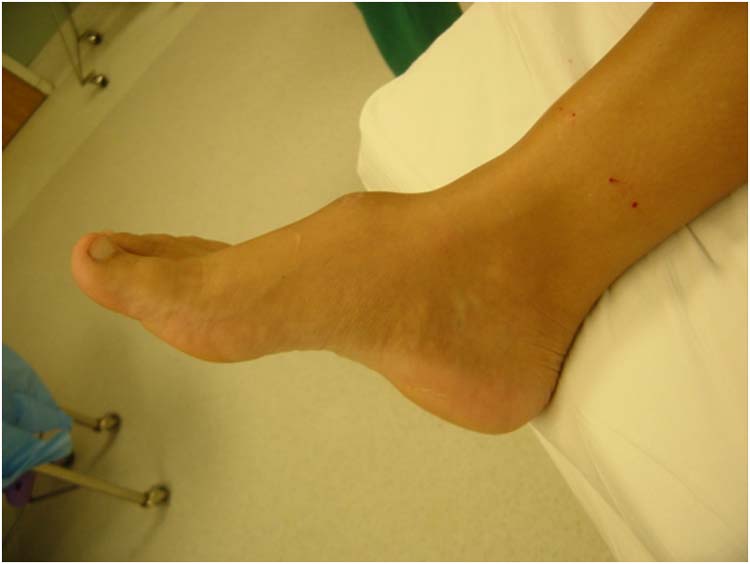 Намного реже можно заметить формы с отклонением от нормы аксонов — объектов осевого направления, которые проходят в центральной части нервной структуры. Аномальные процесс способны повлиять на состояние передних и задних отростков спинного мозга, нейронов передних рогов, путей Голля и столбы Кларка. Повторно из-за проблем с функционированием периферического отдела, начинает образовываться мышечная недостаточность.
Намного реже можно заметить формы с отклонением от нормы аксонов — объектов осевого направления, которые проходят в центральной части нервной структуры. Аномальные процесс способны повлиять на состояние передних и задних отростков спинного мозга, нейронов передних рогов, путей Голля и столбы Кларка. Повторно из-за проблем с функционированием периферического отдела, начинает образовываться мышечная недостаточность.
Разновидности и классификация
В неврологическом направлении медицины невральная амиотрофия Шарко-Мари-Тута разделена на две четких группы, которые клинически достаточно схожи между собой, но имеют список особенностей, позволяющих провести подобное разграничение:
- Невральная амиотрофия I типа — вызывает снижение скорости проведения нервного импульса.
- НА II типа — скорость подвергается пагубному влиянию в меньшей доли, проявляется отклонение нейрита.
В чем замечены проявления невральной амиотрофии Шарко-Мари-Тута?
При описываемом заболевании у человека первым делом берет начало формирование схожих мышечных некрозов в нижних конечностей.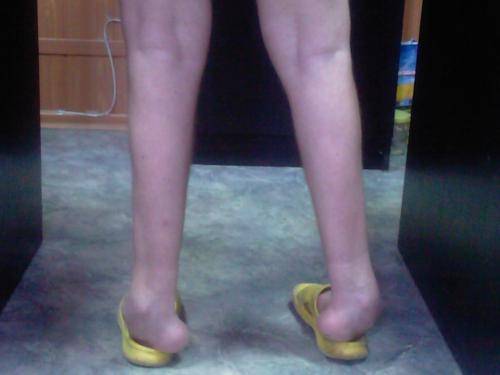 Первые проявления, как правило, происходят такое в возрасте двадцати лет (намного реже, врачи могут диагностировать симптоматику в подростковый период от 16 лет, а также до 30 лет). Подобные проявления состоят в основном в высокой степени утомляемости ног во время долгого нахождения в вертикальном положении. Замечен синдром так называемого «топтания», то есть для того, чтобы пациент мог снять дискомфортные ощущения, он начинает ходить на месте. В некоторых редких ситуациях НА проявляется расстройстве чувствительности тех же отделов, чаще всего — образуются парестезии в виде ползания мурашек.
Первые проявления, как правило, происходят такое в возрасте двадцати лет (намного реже, врачи могут диагностировать симптоматику в подростковый период от 16 лет, а также до 30 лет). Подобные проявления состоят в основном в высокой степени утомляемости ног во время долгого нахождения в вертикальном положении. Замечен синдром так называемого «топтания», то есть для того, чтобы пациент мог снять дискомфортные ощущения, он начинает ходить на месте. В некоторых редких ситуациях НА проявляется расстройстве чувствительности тех же отделов, чаще всего — образуются парестезии в виде ползания мурашек.
Типичным ранних звоночком ШМ, считается полное отсутствие ахилловых и коленных рефлексов сухожилий. В итоге у больного проявляется свисание свода, неспособность ходить на пятках и анормальная походка, которая схожа с лошадиной. Впоследствии пагубный процесс увеличивает степень прогрессирования дальше и затрагивает мышцы и сгибатели. Максимальная уровень омертвения может привести к полной деформации ног с высоким сводом, схожу на тип стопы Фридрейха. Постепенно ШМТ переходит на проксимальные участки, к которым относятся голени и нижние зоны бедер. У пациента начинают серьезные деформации: свисающие стопы, ноги становятся нестандартной формы. Далее поражаются руки, кисть становится похожа на обезьянью лапу.
Постепенно ШМТ переходит на проксимальные участки, к которым относятся голени и нижние зоны бедер. У пациента начинают серьезные деформации: свисающие стопы, ноги становятся нестандартной формы. Далее поражаются руки, кисть становится похожа на обезьянью лапу.
Стоит знать, что пагубные поражения никогда не поражают мышцы шейного периметра, туловища и плечевого района. Помимо вышеперечисленных симптомов, у больного могут появиться следующие проблемы:
- Слабое подергивание.
- Гипертрофия компенсаторной природы мышечных областей.
- Сенсорные нарушения.
- Возможность появления цианоза и отечности на кожном покрове.
Для ШМТ типично медленное развитие симптоматики. Период развития процесса уменьшения в размерах ног и рук может занять до десяти лет. Даже при таких серьёзных деформациях тела больной долгое время способен сохранять работоспособность и нормально выполнять различные бытовые обязанности. Ускорителями симптомов могут стать следующие факторы:
- Попадание в тело инфекционных агентов.
- Долгое пребывание на холоде.
- Травмирования головы.
- Повреждения спины и спинного мозга.
- Нехватка витаминов в организме.
Варианты диагностики
Медицинские работники для диагностирования НА Шарко-Мари-Тута опираются на такие проявления, как:
- Возраст, в котором появились первые признаки недуга.
- Стандартная клиническая картина.
- Схожий характер разрушения организма.
- Медленная степень развития атрофий.
- Увеличение опасности симптомов.
Во время посещения кабинета специалиста, человеку проведут полноценный осмотр тела, чтобы выявить наличие мышечной слабости в стопах и голенях, изменения, отсутствие или серьезное снижение уровня рефлекторных способностей ахиллова и коленного отдела, гипестезию. Для того, чтобы отличить ШМТ от других нервно-мышечных расстройств врачи составляют ряд обследований, которые потребуется пройти инфицированному. К ним относятся:
- Электромиографическое исследование.
- Электронейрография.
- Анализ кровеносных телец на сахар, гормональную норму и наркотические вещества.
- Проведение консультации у специалиста в области генетики.
- ДНК-оценка или секвенирование генома (последний вариант очень дорогостоящий для широкого применения).
- Биопсия.
Способы лечения
В настоящее время медики и ученые всего мира не смогли разработать радикальные варианты избавления от описываемого заболевания. Поэтому для снижения неприятных ощущений у людей с ШМТ применяется симптоматические методики. Больному делают курсы внутримышечного введения витаминов группы В и Е. Для повышения состояния трофики используется АТФ и прочие вещества. Пациентам предписываются ингибиторные препараты холинэстеразы, фармакологические средства для повышения микроциркуляции, особый вид кислот и прочие лекарства. Помимо разных медикаментов больным рекомендуется проходить физтерапевтические процедуры. К ним относятся:
- Электрофорез — при которой организм человека подвергается влиянию электрических сигналов.
- Амплипульстерапия.
- Диадинамотерапия
- Грязелечение.
- Ультразвуковое лечение.
- Оксигенобаротерапия.
- Водолечение в сероводородных, сульфидных, хвойных, радоновых ваннах.

Большое значение для выздоровления человека носит сохранение регулярных двигательных нагрузок, предотвращение формирования изменений и прочих аномалий.
Именно для такого используется ЛФК и массажные сеансы.
При острой необходимости врач-ортопед может предписывать разные ортопедические приемы.
Фридрейх Атаксия Информационный бюллетень | Национальный институт неврологических расстройств и инсульта
Что такое атаксия Фридрейха?
Атаксия Фридрейха (также называемая ФА) — редкое наследственное заболевание, которое вызывает прогрессирующее повреждение нервной системы и проблемы с движением. Обычно это начинается в детстве и приводит к нарушению координации мышц (атаксии), которое со временем ухудшается.
При атаксии Фридрейха нервные волокна спинного мозга и периферических нервов дегенерируют, истончаются. Периферические нервы несут информацию от мозга к телу и от тела обратно к мозгу, такую как сообщение о том, что ступни холодные, или сигнал мышцам для создания движения. Мозжечок, часть мозга, которая координирует баланс и движение, также деградирует в меньшей степени. Это повреждение приводит к неловким, неустойчивым движениям и нарушению сенсорных функций. Заболевание также вызывает проблемы с сердцем (у одной трети заболевших) и позвоночником, а у некоторых людей с этим заболеванием также разовьется диабет.Расстройство не влияет на мышление и мыслительные способности (когнитивные функции).
Периферические нервы несут информацию от мозга к телу и от тела обратно к мозгу, такую как сообщение о том, что ступни холодные, или сигнал мышцам для создания движения. Мозжечок, часть мозга, которая координирует баланс и движение, также деградирует в меньшей степени. Это повреждение приводит к неловким, неустойчивым движениям и нарушению сенсорных функций. Заболевание также вызывает проблемы с сердцем (у одной трети заболевших) и позвоночником, а у некоторых людей с этим заболеванием также разовьется диабет.Расстройство не влияет на мышление и мыслительные способности (когнитивные функции).
Атаксия Фридрейха вызвана дефектом (мутацией) в гене, обозначенном FXN , который несет генетический код белка, называемого фратаксином. У людей, унаследовавших две дефектные копии гена, по одной от каждого родителя, разовьется болезнь. Атаксия Фридрейха, хотя и встречается редко, является наиболее распространенной формой наследственной атаксии в Соединенных Штатах, поражая примерно 1 из 50 000 человек. Дети мужского и женского пола могут унаследовать заболевание.
Дети мужского и женского пола могут унаследовать заболевание.
Скорость прогрессирования варьируется от человека к человеку. Обычно в течение 10-20 лет после появления первых симптомов человек прикован к инвалидной коляске. Люди могут стать полностью недееспособными на более поздних стадиях болезни. Атаксия Фридрейха может сократить продолжительность жизни, а сердечные заболевания являются наиболее частой причиной смерти. Однако некоторые люди с менее серьезными признаками ФА доживают до шестидесяти лет и старше.
Заболевание названо в честь Николауса Фридрейха, немецкого врача, описавшего это заболевание в 1860-х годах.
верх
Каковы признаки и симптомы?
Симптомы обычно появляются в возрасте от 5 до 15 лет, хотя иногда они появляются и в зрелом возрасте. Приблизительно у 15 процентов людей с атаксией Фридрейха наступает атаксия после 25 лет. Первым неврологическим симптомом обычно является затруднение при ходьбе и нарушение равновесия (атаксия походки, часто описываемая как появление головокружения или даже пьянства). Еще один ранний признак заболевания — медлительность и невнятность речи (дизартрия). Со временем речь становится неуверенной и отрывистой (часто называемой «сканированием речи»). Сложность координации движений (атаксия) может повлиять на все мышцы. Он постепенно ухудшается и медленно распространяется на руки и туловище (туловище). По мере прогрессирования мышечной слабости у большинства пораженных людей повышается мышечный тонус (спастичность). Почти у двух третей людей с атаксией Фридрейха также развивается сколиоз (искривление позвоночника в одну сторону), для лечения которого часто требуется хирургическое вмешательство.У большинства пораженных людей также возникают трудности с глотанием из-за нарушения координации мышц языка и горла.
Еще один ранний признак заболевания — медлительность и невнятность речи (дизартрия). Со временем речь становится неуверенной и отрывистой (часто называемой «сканированием речи»). Сложность координации движений (атаксия) может повлиять на все мышцы. Он постепенно ухудшается и медленно распространяется на руки и туловище (туловище). По мере прогрессирования мышечной слабости у большинства пораженных людей повышается мышечный тонус (спастичность). Почти у двух третей людей с атаксией Фридрейха также развивается сколиоз (искривление позвоночника в одну сторону), для лечения которого часто требуется хирургическое вмешательство.У большинства пораженных людей также возникают трудности с глотанием из-за нарушения координации мышц языка и горла.
Помимо нарушения движений, часто наблюдается потеря чувствительности в руках и ногах, которая может распространяться на другие части тела. Другие особенности включают потерю нормальных рефлексов, особенно в коленях и лодыжках, и мышечную слабость. У многих людей с более поздними стадиями атаксии Фридрейха также развивается потеря слуха и зрения.
У многих людей с более поздними стадиями атаксии Фридрейха также развивается потеря слуха и зрения.
Другие симптомы, которые могут возникнуть, включают учащенное сердцебиение и одышку.Эти симптомы являются результатом различных форм сердечных заболеваний, которые часто сопровождают атаксию Фридрейха, таких как увеличение сердца (гипертрофическая кардиомиопатия), образование волокнообразного материала в мышцах сердца (фиброз миокарда) и сердечная недостаточность. Также распространены нарушения сердечного ритма, такие как учащенное сердцебиение (тахикардия) и нарушение проведения сердечных импульсов внутри сердца (блокада сердца).
Около 50 процентов людей с ЖК развиваются непереносимость углеводов, а у 30 процентов развивается диабет.Большинство людей, страдающих этим заболеванием, очень быстро устают и обнаруживают, что им требуется больше отдыха и больше времени, чтобы вылечиться от обычных болезней, таких как простуда и грипп.
верх
Как диагностируется атаксия Фридрейха?
Диагноз атаксии Фридрейха требует тщательного клинического обследования, которое включает в себя сбор анамнеза и тщательный медицинский осмотр, в частности, на предмет нарушения равновесия, потери чувствительности суставов (проприоцепции), отсутствия рефлексов и признаков неврологических проблем. Генетическое тестирование теперь позволяет поставить окончательный диагноз. Другие тесты, которые могут помочь в диагностике или лечении расстройства, включают:
Генетическое тестирование теперь позволяет поставить окончательный диагноз. Другие тесты, которые могут помочь в диагностике или лечении расстройства, включают:
- электромиограмма (ЭМГ), измеряющая электрическую активность мышечных клеток,
- исследования нервной проводимости, которые измеряют скорость, с которой нервы передают импульсы,
- электрокардиограмма (также называемая ЭКГ или ЭКГ), которая дает графическое представление об электрической активности или структуре сердечных сокращений,
- эхокардиограмма, которая фиксирует положение и движение сердечной мышцы,
- анализов крови для проверки на повышенный уровень глюкозы и витамина Е и
- магнитно-резонансная томография (МРТ) или компьютерная томография (КТ), тесты, которые предоставляют изображения головного и спинного мозга, которые полезны для исключения других неврологических состояний.
верх
Как наследуется атаксия Фридрейха?
У людей есть две копии каждого гена, по одной копии наследуются от каждого родителя.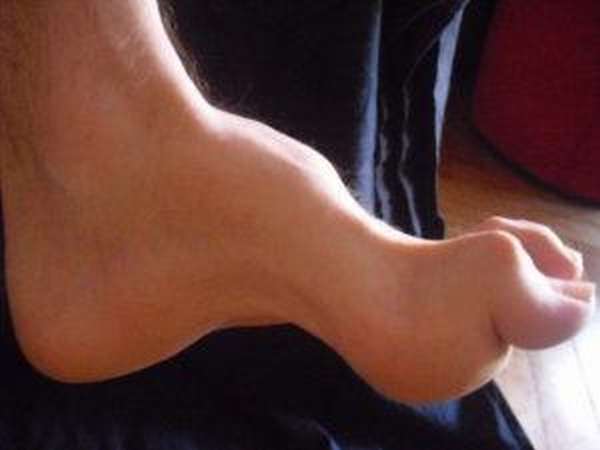 При атаксии Фридрейха человеку необходимо унаследовать две копии дефектного гена FXN для развития болезни. Человек, унаследовавший только одну аномальную копию гена, называется носителем. Носитель не заболеет, но может передать мутацию гена своим детям. Примерно один из 90 американцев европейского происхождения несет аномальный ген FXN .
При атаксии Фридрейха человеку необходимо унаследовать две копии дефектного гена FXN для развития болезни. Человек, унаследовавший только одну аномальную копию гена, называется носителем. Носитель не заболеет, но может передать мутацию гена своим детям. Примерно один из 90 американцев европейского происхождения несет аномальный ген FXN .
верх
Как влияет протеин фратаксин?
Ген FXN предоставляет инструкции по производству белка, называемого фратаксином. В нормальной версии гена триплетная последовательность ДНК (помеченная как гуанин-аденин-аденин или GAA) повторяется от 7 до 22 раз. В дефектном гене FXN повторение GAA повторяется снова и снова — сотни, даже до тысячи раз. Повторяющаяся последовательность GAA значительно снижает количество фратаксина, продуцируемого клеткой.Более раннее начало заболевания и тяжесть прогрессирования могут быть связаны с количеством копий GAA в индивидуальном генетическом коде.
Этот аномальный паттерн, называемый экспансией триплетных повторов, считается причиной нескольких заболеваний, при которых индивидууму необходимо унаследовать только один аномальный ген.![]() Атаксия Фридрейха — единственное известное генетическое заболевание, которое требует наследования двух копий аномального гена FXN , чтобы вызвать заболевание. Почти все люди с FA (98 процентов) имеют две копии этой мутантной формы FXN , но не во всех случаях заболевания.Около двух процентов пораженных людей имеют другие дефекты в гене FXN , которые вызывают заболевание.
Атаксия Фридрейха — единственное известное генетическое заболевание, которое требует наследования двух копий аномального гена FXN , чтобы вызвать заболевание. Почти все люди с FA (98 процентов) имеют две копии этой мутантной формы FXN , но не во всех случаях заболевания.Около двух процентов пораженных людей имеют другие дефекты в гене FXN , которые вызывают заболевание.
Расширение триплетных повторов сильно нарушает нормальную продукцию фратаксина. Фратаксин содержится в вырабатывающих энергию частях клетки, называемых митохондриями. Исследования показывают, что без нормального уровня фратаксина определенные клетки в организме (особенно клетки периферических нервов, спинного мозга, мозга и сердечной мышцы) вырабатывают энергию менее эффективно, и было выдвинуто предположение, что в них накапливаются токсичные побочные продукты, приводящие к тому, что называется « окислительный стресс.Недостаток нормального уровня фратаксина также может привести к повышению уровня железа в митохондриях. Когда избыток железа вступает в реакцию с кислородом, могут образовываться свободные радикалы. Хотя свободные радикалы являются важными молекулами в метаболизме организма, они также могут разрушать клетки и наносить вред организму.
Когда избыток железа вступает в реакцию с кислородом, могут образовываться свободные радикалы. Хотя свободные радикалы являются важными молекулами в метаболизме организма, они также могут разрушать клетки и наносить вред организму.
верх
Можно ли вылечить или вылечить атаксию Фридрейха?
Как и в случае со многими дегенеративными заболеваниями нервной системы, в настоящее время нет лекарства или эффективного лечения атаксии Фридрейха.Однако многие симптомы и сопутствующие осложнения можно лечить, чтобы помочь людям поддерживать оптимальное функционирование как можно дольше. Для лечения человека с атаксией Фридрейха очень важен многопрофильный командный подход. Врачи могут назначить лечение диабета, если таковое имеется; Некоторые проблемы с сердцем также можно лечить с помощью лекарств. Ортопедические проблемы, такие как деформация стопы и сколиоз, можно исправить с помощью скоб или хирургического вмешательства. Физиотерапия может продлить использование рук и ног. Следует внимательно следить за проблемами глотания и речи. С нарушением слуха можно избавиться с помощью слуховых аппаратов.
Следует внимательно следить за проблемами глотания и речи. С нарушением слуха можно избавиться с помощью слуховых аппаратов.
верх
Какие услуги полезны пациентам с атаксией Фридрейха и их семьям?
Генетическое тестирование важно для правильного клинического диагноза и может помочь в пренатальной диагностике и определении статуса носителя. Консультанты-генетики могут помочь объяснить, как передается атаксия Фридрейха.
Врач первичной медико-санитарной помощи может обследовать людей на предмет таких осложнений, как сердечные заболевания, диабет и сколиоз, и может направить людей к таким специалистам, как кардиологи, физиотерапевты и логопеды, чтобы они помогли справиться с некоторыми другими сопутствующими проблемами.
Поддержка и информация для семей также доступна через ряд частных организаций. Эти группы могут предлагать способы создания сетей и общения с другими людьми, затронутыми FA. Они также могут предоставить доступ к реестрам пациентов, информации о клинических испытаниях и другим полезным ресурсам.
верх
Какие исследования проводятся?
Миссия Национального института неврологических расстройств и инсульта (NINDS) состоит в том, чтобы получить фундаментальные знания о мозге и нервной системе и использовать эти знания для уменьшения бремени неврологических заболеваний.NINDS является составной частью Национального института здоровья (NIH), ведущего сторонника биомедицинских исследований в мире.
Диапазон исследований атаксии Фридрейха (FA), финансируемых Национальным институтом здравоохранения, включает определение того, что вызывает мутацию гена и как он функционирует, получение лучшего понимания фратаксина и изучение способов преодоления генетической мутации и разработки методов лечения болезни. В дополнение к NINDS, несколько других институтов и центров NIH поддерживают исследования атаксии Фридрейха.
При атаксии Фридрейха расширенный триплетный повтор GAA снижает выработку фратаксина, но точный механизм того, как ген «заглушается» (выключается), неизвестен. Среди текущих проектов, финансируемых NINDS, исследователи надеются определить механизмы, участвующие в подавлении гена FXN , которые могут выявить потенциальные способы восстановления нормальной функции гена. В одном из таких проектов в качестве модельной системы используются линии индуцированных плюрипотентных стволовых клеток (ИПСК), которые были превращены в нейрональные клетки (действие, называемое , производное ), для изучения механизмов подавления гена FXN .(ИПСК представляют собой тип стволовых клеток, которые могут быть получены из клеток кожи или крови и активированы, чтобы стать другими типами клеток тела.) Надеемся, что эта работа откроет новые терапевтические стратегии для лечения атаксии Фридрейха и, возможно, также связанных с ней заболеваний повторной экспансии. Другой проект использует клеточные модели, полученные из ИПСК, для выявления изменений экспрессии генов при атаксии Фридрейха, чтобы лучше понять митохондриальные дефекты, связанные с заболеванием, и разработать биомаркеры (признаки, которые могут указывать на диагноз или прогрессирование заболевания) для будущих клинических испытаний.
Среди текущих проектов, финансируемых NINDS, исследователи надеются определить механизмы, участвующие в подавлении гена FXN , которые могут выявить потенциальные способы восстановления нормальной функции гена. В одном из таких проектов в качестве модельной системы используются линии индуцированных плюрипотентных стволовых клеток (ИПСК), которые были превращены в нейрональные клетки (действие, называемое , производное ), для изучения механизмов подавления гена FXN .(ИПСК представляют собой тип стволовых клеток, которые могут быть получены из клеток кожи или крови и активированы, чтобы стать другими типами клеток тела.) Надеемся, что эта работа откроет новые терапевтические стратегии для лечения атаксии Фридрейха и, возможно, также связанных с ней заболеваний повторной экспансии. Другой проект использует клеточные модели, полученные из ИПСК, для выявления изменений экспрессии генов при атаксии Фридрейха, чтобы лучше понять митохондриальные дефекты, связанные с заболеванием, и разработать биомаркеры (признаки, которые могут указывать на диагноз или прогрессирование заболевания) для будущих клинических испытаний.
Другие исследователи, поддерживаемые NINDS, работают над созданием новых моделей атаксии Фридрейха на животных, которые точно имитируют генные мутации, обнаруживаемые у людей, пораженных этим заболеванием. Эти новые модели, наряду с уже существующими моделями, необходимы в качестве важнейших инструментов исследования для более детального определения клеточных дефектов болезни и для расширения поиска новых методов лечения.
Исследователи, финансируемые Национальным институтом здравоохранения (NIH), также изучают метаболические дефекты митохондрий (производящих энергию «электростанций» в клетках) у людей с атаксией Фридрейха.Например, один проект анализирует роль фратаксина в биосинтезе железо-серных кластеров в митохондриях. Кроме того, проект программы «Терапия редких и забытых болезней» Национального центра развития трансляционных наук (NCATS) направлен на разработку белковой заместительной терапии атаксии Фридрейха, которая использует новую технологию для доставки функционального белка фратаксина в митохондрии.
Дополнительную информацию об исследованиях атаксии Фридрейха при поддержке NINDS и других институтов и центров NIH можно найти в NIH RePORTER, базе данных с возможностью поиска текущих и прошлых исследовательских проектов, поддерживаемых NIH и другими федеральными агентствами.В RePORTER также есть ссылки на публикации и ресурсы из этих проектов. Еще один полезный ресурс — это ClinicalTrials.gov, реестр всех зарегистрированных клинических испытаний болезней человека, проведенный Национальным институтом здравоохранения.
верх
Где я могу получить дополнительную информацию?
Для получения дополнительной информации о неврологических расстройствах или исследовательских программах, финансируемых Национальным институтом неврологических расстройств и инсульта, свяжитесь с Институтом мозговых ресурсов и информационной сети (BRAIN) по телефону:
МОЗГ
стр.О. Box 5801
Bethesda, MD 20824
800-352-9424
Информацию также можно получить в следующих организациях:
Альянс Фридрейха по исследованию атаксии (FARA)
P.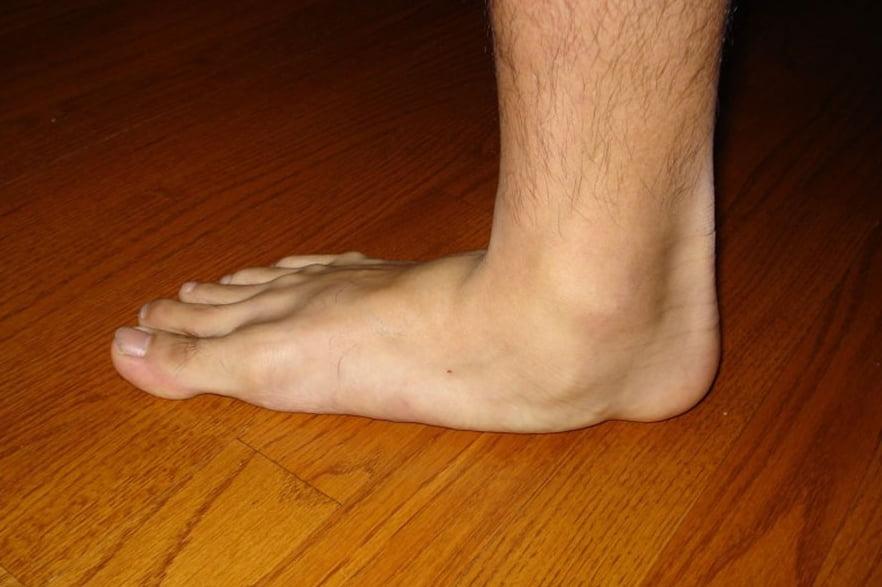 O. Box 1537
O. Box 1537
Springfield, VA 22151
fara@CureFA.org
Tel: 703-426-1576
Fax: (703) 425-0643
Genetic Alliance
4301 Connecticut Avenue, N.W.
Suite 404
Вашингтон, округ Колумбия 20008-2369
info@geneticalliance.org
Тел .: 202-966-5557; 800-336-GENE (4363)
Факс: 202-966-8553
Ассоциация мышечной дистрофии
Национальный офис — 222 S.Riverside Plaza
Suite 1500
Chicago, IL 60606
mda@mdausa.org
Тел .: 800-572-1717
Факс: 520-529-5300
Национальный фонд Атаксии (NAF)
2600 Fernbrook Lane North
Suite 119
Minneapolis, MN 55447-4752
naf@ataxia.org
Тел .: 763-553-0020
Факс: 763-553-0167
Национальная организация по редким заболеваниям (NORD)
55 Kenosia Avenue
Danbury, CT 06810
orphan@rarediseases.org
Тел .: 203-744-0100; Голосовая почта: 800-999-NORD (6673)
Факс: 203-798-2291
У.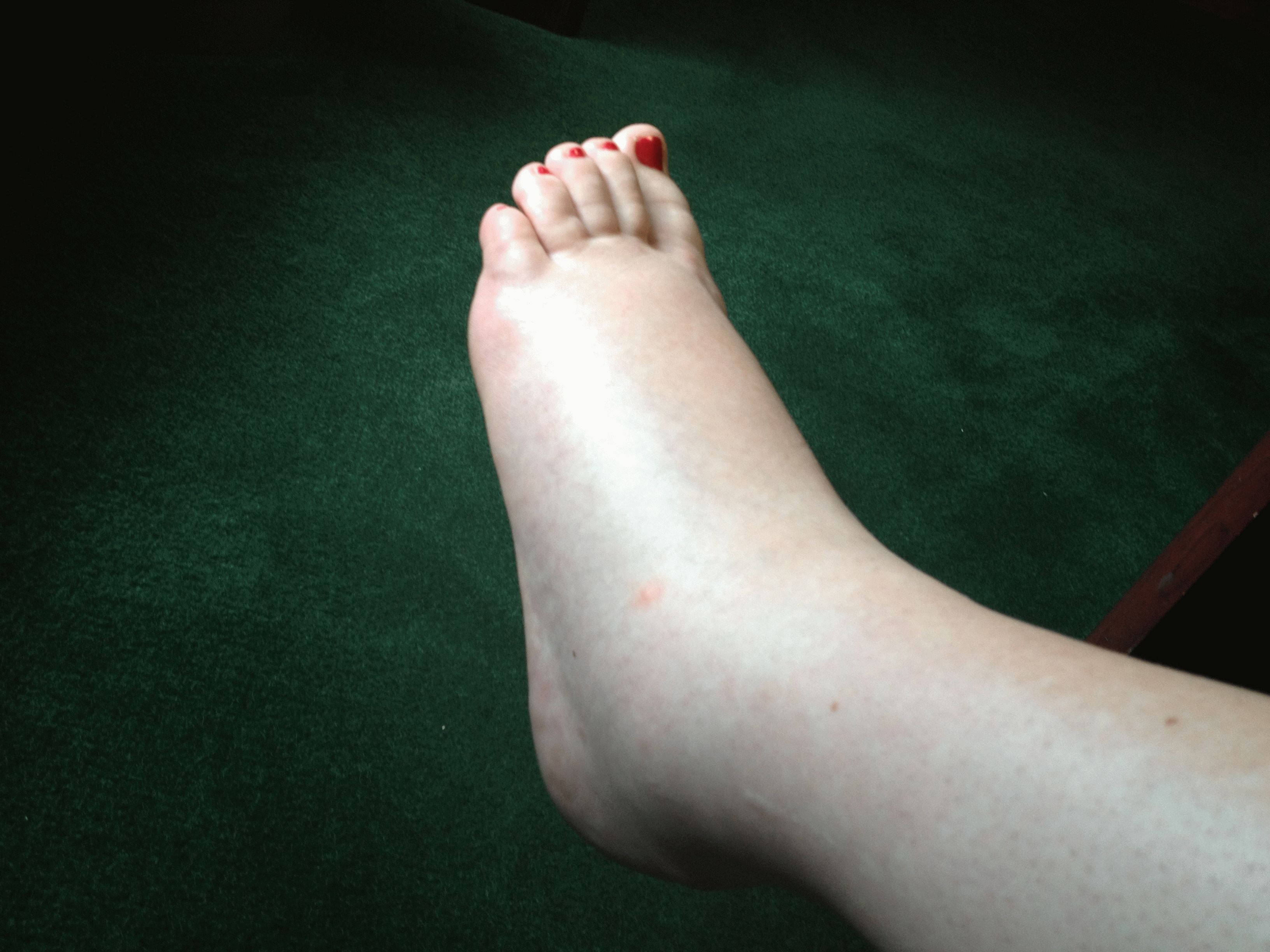 S. Национальная медицинская библиотека
S. Национальная медицинская библиотека
Национальные институты здравоохранения, DHHS
8600 Rockville Pike
Bethesda, MD 20892
888-346-3656
«Информационный бюллетень Фридрейха об атаксии», NINDS, дата публикации июнь 2018 г.
Публикация NIH № 18-NS-87
Назад к странице информации об атаксии Фридрейха
См. Список всех расстройств NINDS
Publicaciones en Español
Ataxia de Friedreich
Подготовлено:
Офис по связям с общественностью
Национальный институт неврологических расстройств и инсульта
Национальные институты здравоохранения
Bethesda, MD 20892
NINDS, связанные со здоровьем, предоставляются только в информационных целях и не обязательно представляют собой одобрение или официальную позицию Национального института неврологических расстройств и инсульта или любого другого федерального агентства.Консультации по лечению или уходу за отдельным пациентом следует получать после консультации с врачом, который обследовал этого пациента или знаком с историей болезни этого пациента.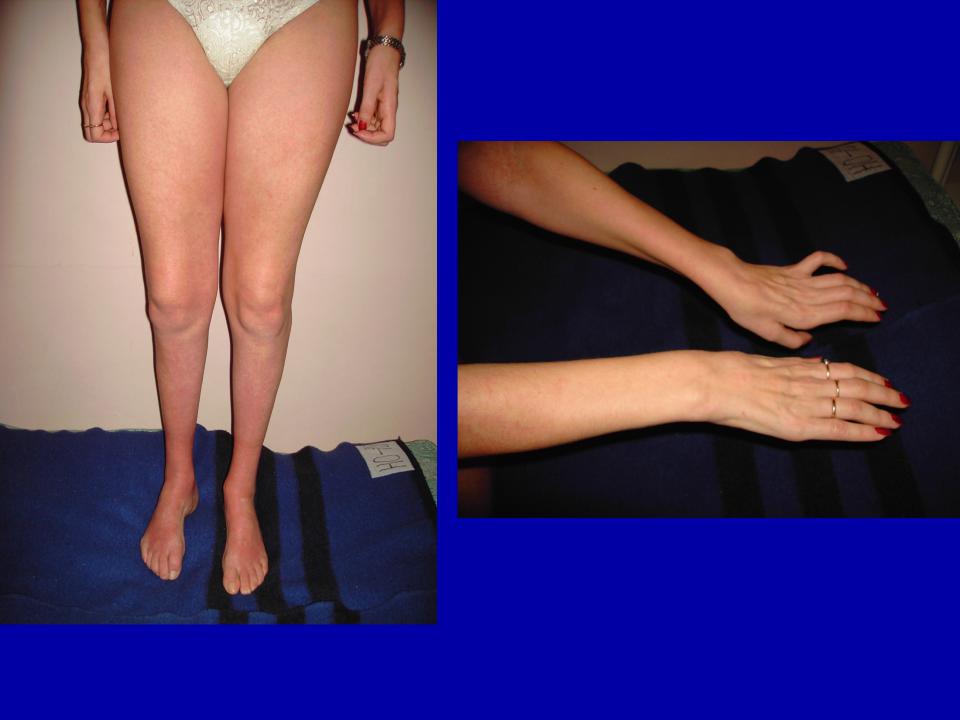
Вся информация, подготовленная NINDS, находится в открытом доступе и может свободно копироваться. Благодарность NINDS или NIH приветствуется.
Атаксия Фридрейха | Педиатрическое ортопедическое общество Северной Америки (POSNA)
Учебное пособие
Ключевые точки:
- Прогрессирующее нервно-мышечное расстройство, затрагивающее в основном сердце и нервную систему
- Клинические признаки включают атаксию, потерю глубоких сухожильных рефлексов в коленном и голеностопном суставах и восходящий симптом Бабинского.
- Ортопедические проблемы включают сколиоз и кавоварусную ногу
- Сопутствующие медицинские заболевания, включая кардиомиопатию и сахарный диабет
Описание:
Атаксия Фридрейха была впервые описана в 1863 году Николаусом Фридрейхом, профессором медицины в Германии.Это аутосомно-рецессивное заболевание, вызванное мутацией в гене FXN, который влияет на образование белка фратаксина.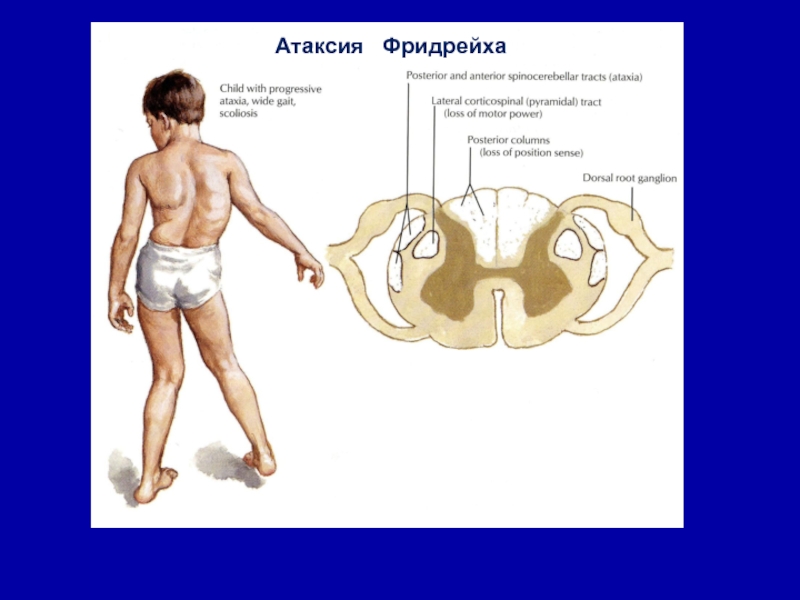 Клинически наблюдается острое начало с прогрессирующим поражением нервной системы. Начало обычно происходит в возрасте от 10 до 15 лет. Последовательность и тяжесть симптомов сильно различаются (Pandolfo, 2009).
Клинически наблюдается острое начало с прогрессирующим поражением нервной системы. Начало обычно происходит в возрасте от 10 до 15 лет. Последовательность и тяжесть симптомов сильно различаются (Pandolfo, 2009).Эпидемиология:
Пострадало около 1/50 000 детей (Pandolfo, 2009). Это наиболее частая форма дегенеративного заболевания спинного мозга.Клинические результаты:
Пациенты с атаксией Фридрейха сначала жалуются на плохую координацию, мышечную слабость или атаксию. При дополнительном обследовании часто наблюдается потеря рефлексов. У этих детей также могут быть сенсорные нарушения, включая снижение чувствительности и, иногда, потерю слуха или зрения.Pes cavus изначально может быть гибким, но может стать фиксированным и потребовать хирургического вмешательства. Снижение проприоцепции также можно отметить при осмотре стопы.
Сколиоз встречается у 63% пациентов (Milbrandt, 2008). Это часто связано с гиперкифозом, и кривые различаются как по типу, так и по величине (Milbrandt, 2008; Tsirikos, 2012).
С медицинской точки зрения следует контролировать гипертрофическую кардиомиопатию (Pandolfo, 2009). Сердечная недостаточность может привести к снижению продолжительности жизни. Около 10% пациентов обращаются с диабетом (Pandolfo, 2009).
Исследования изображений:
Первоначальная визуализация должна быть основана на клинических данных.Рентгенограммы позвоночника часто делают из-за высокой степени сколиоза. Если есть деформация стопы, получают соответственно переднюю и боковую часть стопы или ступни.Этиология:
Атаксия Фридрейха — это аутосомно-рецессивное заболевание, которое возникает в результате мутации гена FXN на хромосоме 9, что приводит к снижению экспрессии белка фратаксина (Pandolfo, 2009).Обращение:
Существуют различные виды лечения, которыми обычно не занимаются хирурги-ортопеды. Что касается ортопедических вмешательств, иногда рекомендуется коррекция симптоматической кавусной мышцы. Это может повлечь за собой расщепление подошв, перенос сухожилий и / или остеотомию плюсневой кости и пяточной кости.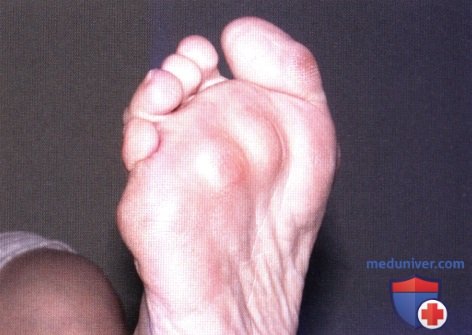 Если пациент обращается на поздних стадиях заболевания или не требует амбулаторного лечения, тройной артродез может быть лучшим хирургическим вмешательством.
Если пациент обращается на поздних стадиях заболевания или не требует амбулаторного лечения, тройной артродез может быть лучшим хирургическим вмешательством.
Ортезы сколиоза редко эффективны для предотвращения прогрессирования искривления (Milbrandt, 2008; Tsirikos, 2012). Спондилодез рекомендуется при изгибе более 50–60 градусов (Milbrandt, 2008; Tsirikos, 2012).Следует отметить, что у этих пациентов мониторинг SSEP часто оказывается неэффективным (Milbrandt, 2008). Оперирующему хирургу может потребоваться пробуждение.
Осложнения:
Дисфункция сердца является частой причиной преждевременной смерти пациентов с атаксией Фридрейха (Tsirikos, 2012; Pandolfo, 2009). Прогрессирующая атаксия и мышечная слабость часто требуют использования инвалидной коляски для передвижения и необходимости помощи в повседневной деятельности. Дизартрия — обычное явление (Pandolfo, 2009).Артикул:
- Милбрандт Т.А., Кунес-младший, Кароль Л.А. Атаксия и сколиоз Фридрейха: опыт двух медицинских учреждений. JPO, 2008. 28 (2): стр. 234-8.
- Атаксия Пандольфо М. Фридрейха: клиническая картина. Журнал неврологии, 2009. Март; 256 приложение 1: с. 3-8.
- Цирикос А.И., Смит Г. Сколиоз у пациентов с атаксией Фридрейха. JBJS Br, 2012. 94 (5): с. 684-9.
 JPO, 2008. 28 (2): стр. 234-8.
JPO, 2008. 28 (2): стр. 234-8.Ведущие участники:
Дана Ольшевски MDАтаксия Фридрейха | Johns Hopkins Medicine
Что такое атаксия Фридрейха?
Атаксия Фридрейха — редкое наследственное дегенеративное заболевание.Он повреждает спинной мозг, периферические нервы и мозжечок. Это состояние имеет тенденцию развиваться у детей и подростков и со временем постепенно ухудшается. По мере прогрессирования болезни развиваются неустойчивые, неловкие движения и потеря чувствительности из-за повреждения нервов.
Состояние названо в честь Николая Фридрейха, немецкого врача, обнаружившего его в 1860-х годах. Атаксия означает нарушение и несогласованность движений мышц, приводящее к дисбалансу походки
Что вызывает атаксию Фридрейха?
Атаксия Фридрейха — это наследственное заболевание, вызванное дефектом гена FXN. Это рецессивное генетическое заболевание. Это означает, что для его развития вам необходимо унаследовать копию дефекта гена от обоих родителей.
Это рецессивное генетическое заболевание. Это означает, что для его развития вам необходимо унаследовать копию дефекта гена от обоих родителей.
Каковы симптомы атаксии Фридрейха?
Симптомы атаксии Фридрейха часто начинаются в возрасте от 5 до 15 лет, хотя они могут развиться и в более позднем возрасте. Физические симптомы включают:
Эти симптомы могут быть вызваны повреждением нервов. Или они могут быть вызваны проблемами с сердцем, которые имеют тенденцию развиваться у людей с этим заболеванием. У многих людей в конечном итоге развивается сколиоз (искривление позвоночника в одну сторону) или деформации стопы, которые часто требуют хирургического вмешательства.У некоторых также развивается диабет.
Симптомы атаксии Фридрейха со временем ухудшаются.
Как диагностируется атаксия Фридрейха?
Чтобы диагностировать атаксию Фридрейха, ваш врач изучит вашу историю болезни и проведет медицинский осмотр. Специальные тесты для диагностики этого состояния могут включать:
Проверка рефлексов, равновесия и нервной чувствительности
Анализы крови
Сканирование изображений, таких как МРТ (магнитно-резонансная томография) или КТ (компьютерная томография)
Исследования нервной проводимости с использованием электрических импульсов для проверки нервной активности
Сердечные пробы
Генетические тесты для выявления дефектного гена, вызывающего заболевание
Как лечится атаксия Фридрейха?
Нет лекарства от атаксии Фридрейха, но некоторые потенциально прорывные методы лечения находятся в стадии изучения.
Лечение направлено на минимизацию симптомов и поддержание комфорта и функциональности как можно дольше. Варианты включают скобы для поддержки рук, ног, ступней или позвоночника; физиотерапия; Логопедия; трудотерапия; и операция по исправлению проблем со скелетом.
Вам могут потребоваться лекарства, если у вас возникнут проблемы с сердцем или диабет.
Жизнь с атаксией Фридрейха
Как и другие дегенеративные заболевания нервной системы, атаксия Фридрейха имеет тенденцию прогрессировать с течением времени, но течение может различаться у разных людей.Лечение часто помогает ограничить симптомы и как можно дольше держать это состояние под контролем.
По мере прогрессирования заболевания оно часто приводит к сколиозу — аномальному искривлению позвоночника — или деформациям стопы, по поводу которых может потребоваться операция.
Он также может вызывать проблемы с сердцем из-за ослабленной сердечной мышцы и нарушений в электрической системе вашего сердца. У некоторых людей с этим заболеванием развивается диабет. Важно следовать советам вашего лечащего врача, чтобы ограничить последствия этих осложнений.
У некоторых людей с этим заболеванием развивается диабет. Важно следовать советам вашего лечащего врача, чтобы ограничить последствия этих осложнений.
Из-за воздействия атаксии Фридрейха на нервную систему и другие органы многие люди должны будут находиться в инвалидном кресле в течение 10–20 лет. По мере прогрессирования болезни также могут возникать невнятная речь, потеря слуха и проблемы со зрением. Некоторые люди могут умереть от сердечной недостаточности или других осложнений. Однако некоторые люди доживают до 70 лет и старше.
Для людей с постоянными физическими нарушениями физиотерапия может быть очень важной для поддержания или увеличения силы и улучшения координации.Другой вид терапии — трудотерапия. Эта терапия учит новым способам выполнения повседневных задач, несмотря на физические ограничения. Логопедия также может быть полезна при нарушении речи.
Наконец, некоторые люди с физическими недостатками могут чувствовать грусть или депрессию. Для лечения депрессии доступны и антидепрессанты, и психотерапия, или «разговорная терапия».
Когда мне следует позвонить своему врачу?
Если вам поставили диагноз атаксия Фридрейха, поговорите со своим врачом о том, когда вам, возможно, придется им позвонить.Скорее всего, они посоветуют вам позвонить, если вы заметите ухудшение каких-либо симптомов или у вас появятся какие-либо новые симптомы.
Ключевые моменты
Атаксия Фридрейха — это наследственное заболевание, поражающее некоторые нервы организма. Это вызвано дефектом гена, который унаследован от обоих родителей.
Симптомы часто начинаются в позднем детстве и могут включать проблемы с ходьбой, утомляемость, изменения чувствительности и замедленную речь. Они имеют тенденцию к ухудшению со временем.Также могут возникать сердечные заболевания, проблемы со скелетом и диабет.
Атаксия Фридрейха в настоящее время не излечима, но сейчас изучаются новые методы лечения. Современные методы лечения, такие как хирургия, физиотерапия, профессиональная терапия и логопедия, направлены на то, чтобы держать болезнь под контролем как можно дольше.
Лекарства часто используются для лечения болезней сердца или диабета.
 Лекарства часто используются для лечения болезней сердца или диабета.
Лекарства часто используются для лечения болезней сердца или диабета.Симптомы атаксии Фридрейха — Новости атаксии Фридрейха
Атаксия Фридрейха (FA) — это генетическое заболевание, вызванное расширением тринуклеотидного повтора GAA в гене FXN, которое поражает нервную систему и мышцы.У лиц с повышенным числом повторов GAA в гене FXN FA развивается раньше и тяжелее, чем у лиц с меньшим числом повторов. Симптомы атаксии Фридрейха обычно проявляются в возрасте от 5 до 15 лет. У людей с низким числом повторов (менее 300) болезнь проявляется поздно (после 25 лет). В редких случаях заболевание начинается в возрасте 75 лет.
Хотя скорость прогрессирования варьирует, человеку с FA необходимо инвалидное кресло в течение 10–20 лет после появления симптомов.На более поздних стадиях болезни люди могут полностью ослабнуть.
FA может сократить продолжительность жизни, при этом сердечные заболевания являются наиболее частой причиной смерти. Однако некоторые люди с менее тяжелым заболеванием могут дожить до шестидесяти, семидесяти или даже старше.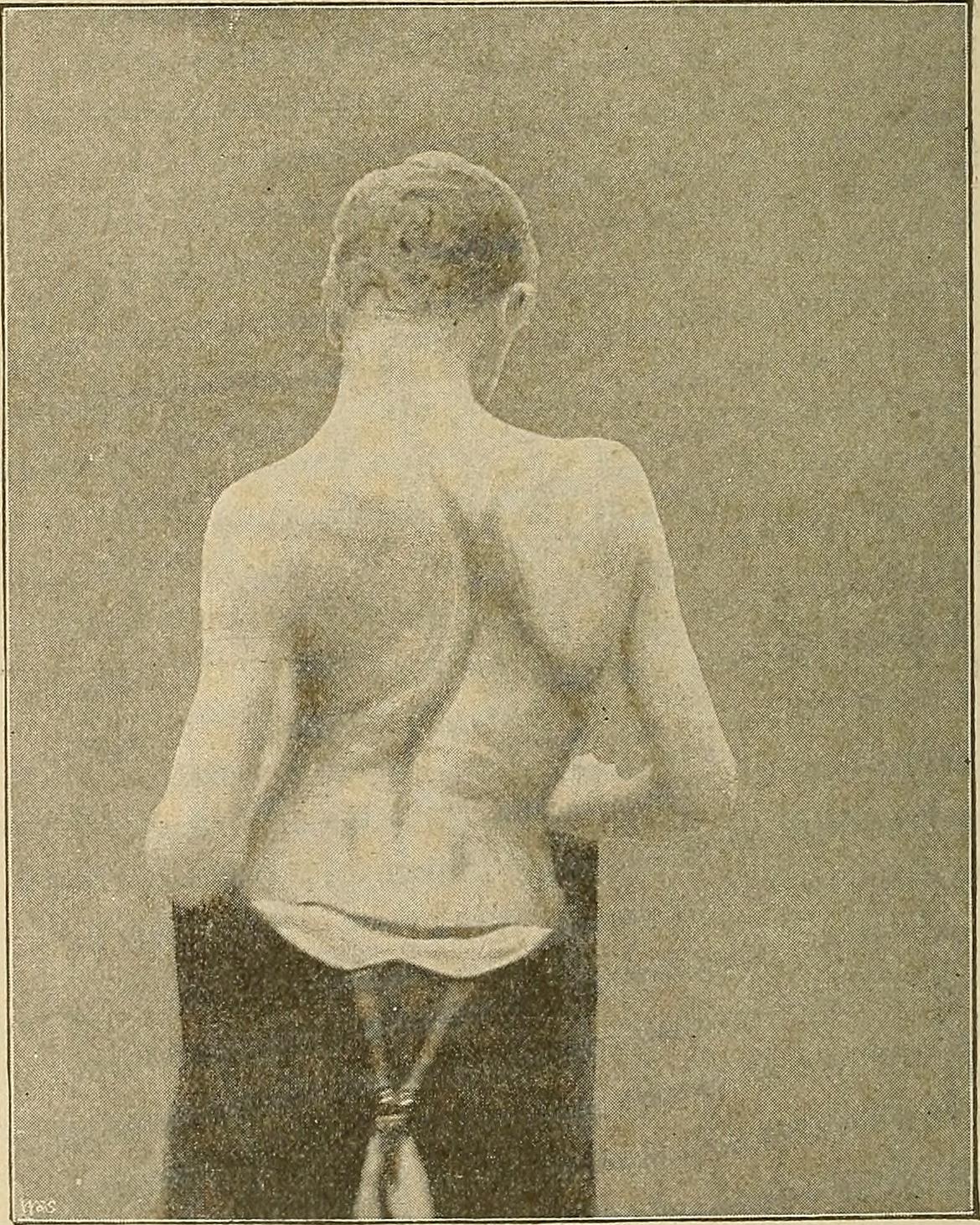
Хотя умственные способности людей с ФА остаются полностью неизменными, прогрессирующая потеря координации и мышечной силы приводит к двигательной инвалидности и необходимости пользоваться инвалидной коляской. Большинству молодых пациентов с FA (подросткам или лицам в возрасте от 20 лет) требуются вспомогательные средства передвижения, такие как трость, ходунки или инвалидное кресло.
FA поражает многие органы и, таким образом, вызывает ряд симптомов.
Нервно-мышечные симптомы К ним относятся потеря координации или атаксия в руках и ногах, сопровождающаяся неуклюжими движениями и тряской. Атаксия походки или затруднения при ходьбе обычно являются первыми симптомами. Это постепенно ухудшается и распространяется на руки и туловище. Другие симптомы могут включать трудности с равновесием, паралич мышц ног, затруднение движений руками и потерю чувствительности (особенно вибрации и ощущения положения) в конечностях.Другие особенности включают потерю рефлексов в коленях и лодыжках.
Деформации скелета вызваны нервно-мышечными проблемами. К ним относятся агрессивный сколиоз (искривление позвоночника в одну сторону), высокий свод стопы (pes cavus), косолапость, деформации пальцев стопы и инверсии стопы (стопы, повернутые внутрь). Сколиоз часто требует хирургического вмешательства.
Неврологические проблемыНеврологические проблемы, связанные с ФА, включают затрудненную речь или невнятную речь, называемые дизартрией.Он включает в себя медленную, отрывистую и нерешительную речь, которая постепенно ухудшается. Другие проблемы включают быстрые, непроизвольные, резкие движения глазных яблок (нистагм), снижение зрения и потерю слуха.
Проблемы с сердцем У 75 процентов людей с ФА развиваются сердечные аномалии. Различные формы сердечных заболеваний, сопровождающих ФА, включают гипертрофическую кардиомиопатию (увеличение и ослабление сердечной мышцы), фиброз миокарда (образование волокнообразного материала в мышцах сердца) и сердечную недостаточность.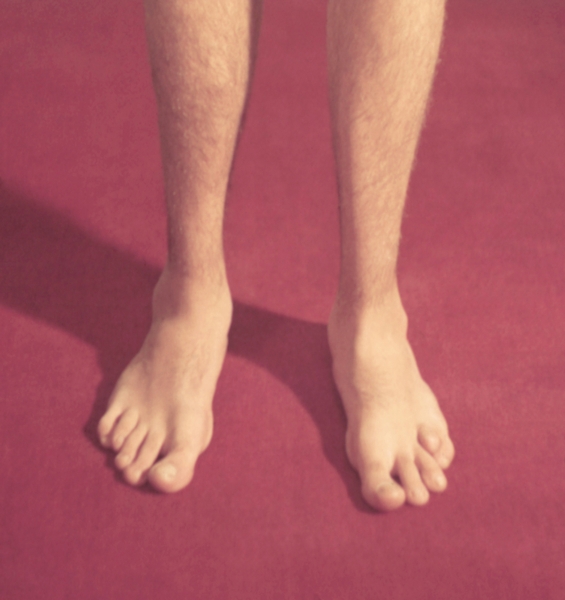 Также распространены нарушения сердечного ритма, такие как тахикардия (учащенное сердцебиение) и блокада сердца (нарушение проведения сердечных импульсов внутри сердца).
Также распространены нарушения сердечного ритма, такие как тахикардия (учащенное сердцебиение) и блокада сердца (нарушение проведения сердечных импульсов внутри сердца).
Эти проблемы с сердцем приводят к таким симптомам, как крайняя усталость, боль в груди, одышка (особенно при физической нагрузке) и ненормально быстрое или нерегулярное сердцебиение (учащенное сердцебиение). Симптомы сердечной недостаточности включают отек ног и затрудненное дыхание в горизонтальном положении.
ДиабетОколо 20 процентов людей с ЖК развиваются непереносимость углеводов, а у 10 процентов развивается диабет.Симптомы диабета включают сильную жажду, частое мочеиспускание, потерю веса, усталость и нечеткое зрение.
Примечание. Friedreich’s Ataxia News — это исключительно новостной и информационный веб-сайт об этой болезни. Он не предоставляет медицинских консультаций, диагнозов или лечения. Этот контент не предназначен для замены профессиональных медицинских консультаций, диагностики или лечения. Всегда обращайтесь за советом к своему врачу или другому квалифицированному поставщику медицинских услуг по любым вопросам, которые могут у вас возникнуть относительно состояния здоровья.Никогда не игнорируйте профессиональные медицинские советы и не откладывайте их поиск из-за того, что вы прочитали на этом веб-сайте.
Всегда обращайтесь за советом к своему врачу или другому квалифицированному поставщику медицинских услуг по любым вопросам, которые могут у вас возникнуть относительно состояния здоровья.Никогда не игнорируйте профессиональные медицинские советы и не откладывайте их поиск из-за того, что вы прочитали на этом веб-сайте.
Атаксия Фридрейха | UF Health, University of Florida Health
Определение
Атаксия Фридрейха — редкое заболевание, передающееся от семьи (наследуемое). Это влияет на мышцы и сердце.
Альтернативные названия
Атаксия Фридрейха; Спиноцеребеллярная дегенерация
Причины
Атаксия Фридрейха вызывается дефектом гена фратаксина (FXN).Изменения в этом гене заставляют организм производить слишком много части ДНК, называемой тринуклеотидным повтором (GAA). Обычно тело содержит от 8 до 30 копий GAA. У людей с атаксией Фридрейха есть целых 1000 копий. Чем больше у человека копий GAA, тем раньше в жизни начинается болезнь и тем быстрее она ухудшается.
Атаксия Фридрейха — аутосомно-рецессивное генетическое заболевание. Это означает, что вы должны получить копию дефектного гена и от матери, и от отца.
Симптомы
Симптомы вызваны стиранием структур в областях головного и спинного мозга, которые контролируют координацию, движение мышц и другие функции.Симптомы чаще всего начинаются до полового созревания. Симптомы могут включать:
Проблемы с мышцами приводят к изменениям в позвоночнике. Это может привести к сколиозу или кифосколиозу.
Чаще всего развивается сердечная недостаточность. Сердечная недостаточность или аритмии, которые не поддаются лечению, могут привести к смерти. Диабет может развиться на более поздних стадиях болезни.
Экзамены и тесты
Можно сделать следующие тесты:
Тесты на сахар (глюкозу) в крови могут показать диабет или непереносимость глюкозы.При осмотре глаза можно выявить повреждение зрительного нерва, которое чаще всего протекает бессимптомно.
Лечение
Лечение атаксии Фридрейха включает:
- Консультации
- Логопедия
- Физиотерапия
- Средства для ходьбы или инвалидные коляски
Ортопедические приспособления (скобы) могут потребоваться при сколиозе и проблемах со стопами. Лечение болезней сердца и диабета помогает людям жить дольше и улучшать качество их жизни.
Лечение болезней сердца и диабета помогает людям жить дольше и улучшать качество их жизни.
Перспективы (Прогноз)
Атаксия Фридрейха постепенно ухудшается и вызывает проблемы при выполнении повседневных дел.Большинству людей требуется инвалидная коляска в течение 15 лет после начала заболевания. Заболевание может привести к преждевременной смерти.
Возможные осложнения
Осложнения могут включать:
- Диабет
- Сердечная недостаточность или болезнь сердца
- Утрата способности передвигаться
Когда обращаться к медицинскому работнику
При появлении симптомов атаксии Фридрейха позвоните своему врачу возникают, особенно если есть семейный анамнез заболевания.
Профилактика
Люди с семейным анамнезом атаксии Фридрейха, которые намереваются иметь детей, могут захотеть рассмотреть возможность генетического скрининга для определения своего риска.
Изображения
Ссылки
Mink JW.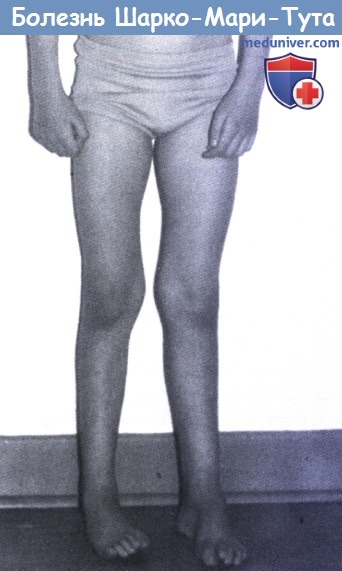 Двигательные расстройства. В: Kliegman RM, Stanton BF, St. Geme JW, Schor NF, ред. Учебник педиатрии Нельсона . 20-е изд. Филадельфия, Пенсильвания: Эльзевьер; 2016: глава 597.
Двигательные расстройства. В: Kliegman RM, Stanton BF, St. Geme JW, Schor NF, ред. Учебник педиатрии Нельсона . 20-е изд. Филадельфия, Пенсильвания: Эльзевьер; 2016: глава 597.
Warner WC, Sawyer JR. Сколиоз и кифоз. В: Azar FM, Beaty JH, Canale ST, ред. Оперативная ортопедия Кэмпбелла . 13-е изд. Филадельфия, Пенсильвания: Эльзевьер; 2017: глава 44.
Атаксия Фридрейха — AMBOSS
Последнее обновление: 12 ноября 2020 г.
Резюме
Атаксия Фридрейха (FDRA) — аутосомно-рецессивное заболевание, включающее расширение тринуклеотидных повторов, которое приводит к прогрессирующей нейродегенерации. Он поражает несколько трактов спинного мозга, вызывая мышечную слабость и нарушение координации всех конечностей. Пошатывающаяся походка в детстве — вот главный симптом.Другие особенности включают аномалии скелета, кардиомиопатию и диабет. Клиническое течение в основном определяется степенью потери подвижности и поражением сердца. Диагноз требует подтверждения с помощью генетических тестов.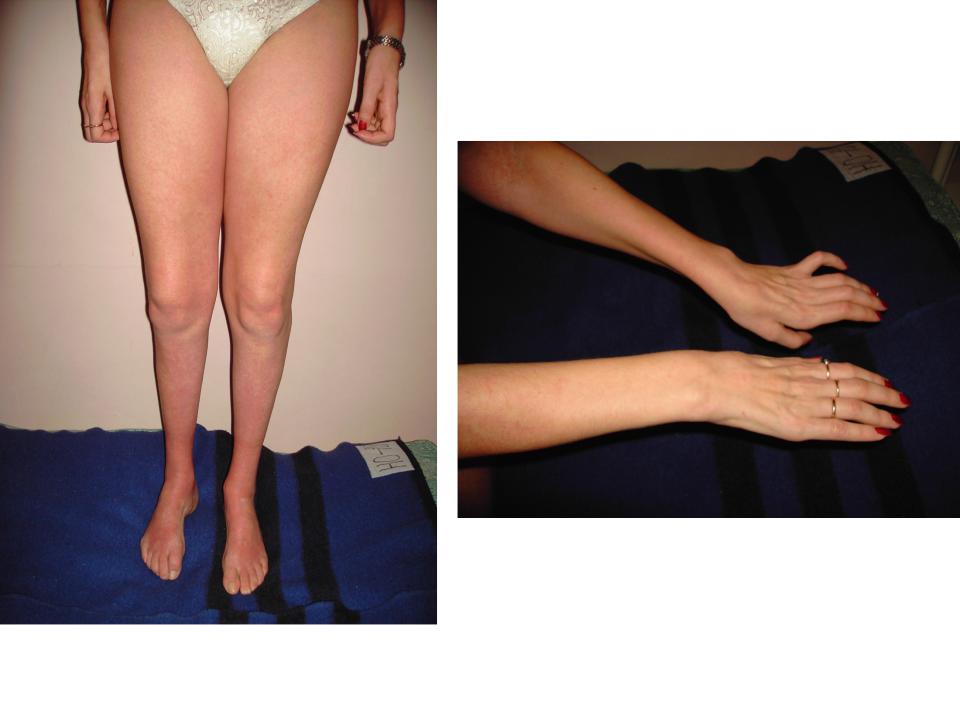 Поскольку лечебной терапии нет, прогноз плохой.
Поскольку лечебной терапии нет, прогноз плохой.
Эпидемиология
Эпидемиологические данные относятся к США, если не указано иное.
Этиология
Ссылки: [2] [4]
Клинические особенности
- Неврологический
- Деформации скелета
- Другие особенности
Лица с FRiedreich ATAXia имеют ATAXic GAAit из-за расширения триплета GAA в гене FRATAXin.
Диагностика
Каталожные номера: [4]
Лечение
- Лечебное лечение недоступно
- Многопрофильная поддерживающая помощь: генетическое консультирование, физиотерапия, речевая и языковая терапия
- Лечение кардиологических, ортопедических и метаболических (например, диабета) особенностей заболевания
Ссылки: [4]
Прогноз
- К возрасту 20–30 лет не ходят.
- Средний возраст смерти: 37 лет
- Причины смерти
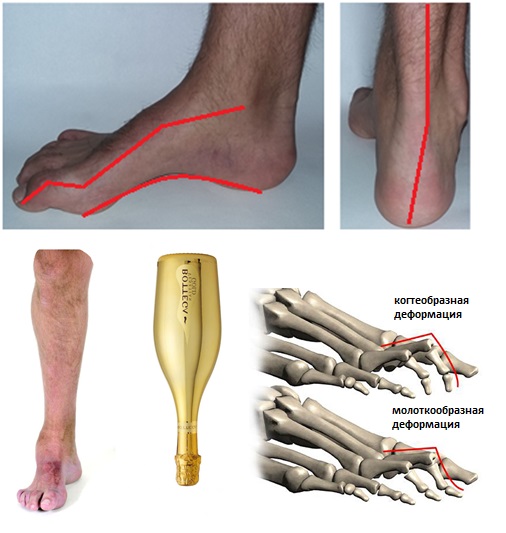
Ссылки: [5]
Профилактика
- Тестирование родственников на носительство
- Пренатальное тестирование: Родителям с одним больным ребенком рекомендуется генетическое консультирование.
Каталожные номера: [5]
Ссылки
- Кук А., Атаксия Джунти П. Фридрейха: клинические особенности, патогенез и лечение. Бр. Мед Булл . 2017; 124 (1): стр.19-30. DOI: 10.1093 / bmb / ldx034. | Открыть в режиме чтения QxMD
- Пандольфо М. Фридрейх Атаксия. Arch Neurol . 2008; 65 (10). DOI: 10.1001 / archneur.65.10.1296. | Открыть в режиме чтения QxMD
- Фридрейх Атаксия. http://www.omim.org/clinicalSynopsis/229300 . Обновлено: 6 октября 2011 г.
Доступ: 3 апреля 2017 г.
- Руано Л., Мело С., Сильва М.К., Коутиньо П. Глобальная эпидемиология наследственной атаксии и спастической параплегии: систематический обзор исследований распространенности. Нейроэпидемиология . 2014; 42 (3): с.174-183. DOI: 10,1159 / 000358801. | Открыть в режиме чтения QxMD
- Майкл Х. Паркинсон, Сильвия Бош, Вольфганг Начбауэр, Катерина Мариотти, Паола Джунти.Клинические особенности атаксии Фридрейха: классические и атипичные фенотипы. Дж. Нейрохим . 2013; 126 : с.103-117. DOI: 10.1111 / jnc.12317. | Открыть в режиме чтения QxMD
 Обновлено: 6 октября 2011 г.
Доступ: 3 апреля 2017 г.
Обновлено: 6 октября 2011 г.
Доступ: 3 апреля 2017 г.Атаксия Фридрейха; FA, FRDA информация и лечение
Синоним: Атаксия Фридрейха
Атаксия Фридрейха является наиболее распространенной наследственной атаксией в Великобритании. Это дегенеративное заболевание, которое в первую очередь поражает нервную систему и сердце. Это часто связано с кардиомиопатией или диабетом. [1]
Это часто связано с кардиомиопатией или диабетом. [1]
Наблюдается прогрессирующая атаксия, дизартрия, снижение проприоцепции / чувствительности к вибрации и мышечная слабость. Наследование аутосомно-рецессивное.
Атаксия Фридрейха была впервые описана в 1863 году немецким врачом Николаусом Фридрейхом. Ген атаксии Фридрейха был открыт в 1996 году, что привело к лучшему распознаванию спектра заболеваний.
Эпидемиология
[2]- Распространенность составляет около 1,8 на 100 000 в Великобритании.
- Атаксия Фридрейха встречается у населения из Европы, Ближнего Востока, Северной Африки и Индии. Он отсутствует в популяциях из Восточной Азии (Китай и Япония) и у американских индейцев.
Этиология и генетика
[3, 4]- Генное заболевание представляет собой мутацию гена фратаксина на хромосоме 9.
- У большинства пациентов (98%) наблюдается экспансия GAA-повтора гена фратаксина; размер расширения коррелирует с тяжестью заболевания. [5]
- Реже точечная мутация встречается в одном гене.
- Мутация приводит к снижению синтеза фратаксина, митохондриального белка.
- Функция белка фратаксина неизвестна, но, по-видимому, он участвует в гомеостазе клеточного железа. Его дисфункция приводит к дисфункции митохондрий и окислительному повреждению клеток. [6]
- Повреждение клеток в первую очередь затрагивает нервную систему и сердце (по неизвестным причинам). Сенсорные нейроны, связанные с проприоцепцией, поражаются рано и серьезно, поэтому атаксия является ранним признаком заболевания. [6]
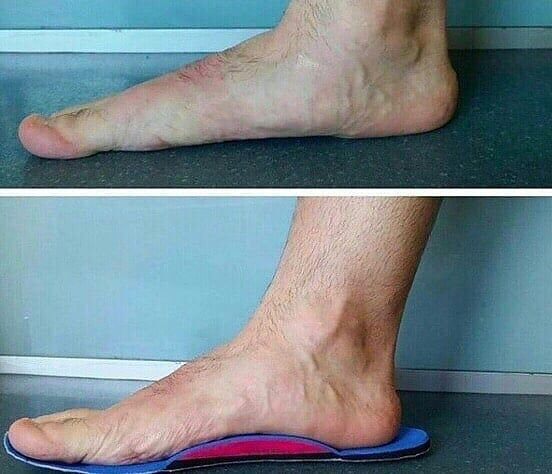 [5]
[5] Клинические особенности
[2, 4] Атаксия Фридрейха — прогрессирующее нейродегенеративное заболевание, обычно начинающееся в возрасте до 20 лет. Признаки и симптомы включают прогрессирующую атаксию, восходящую слабость и восходящую потерю вибрации и чувствительности положения суставов, кавус, сколиоз, кардиомиопатию и сердечную аритмию. [7]
[7]
Начало
- Обычно проявляется до подросткового возраста, с началом в возрасте от 2 до 16 лет.
- Возможно позднее начало заболевания в возрасте до 50 лет с более легкими формами атаксии Фридрейха.
Наличие симптомов
- Неустойчивость походки — обычный симптом.
- Другими симптомами являются общая неуклюжесть или ухудшение спортивных результатов.
- Иногда проявляется сколиозом или каверной стопой, которые могут предшествовать неврологическим симптомам на несколько лет.
- Может проявляться (редко) с симптомами кардиомиопатии.
- В одном описании случая атаксии не было, но с потерей зрения, двигательных функций и чувствительности. [8]
Неврологические особенности
- Атаксия в основном является результатом дегенерации нейронов ганглиев задних корешков, что приводит к афферентной атаксии.
- Поражение черепных нервов может вызвать дизартрию, потерю зрения и слуха, а также дисфагию.
- Неврологические признаки:
- Прогрессирующая атаксия конечностей и походки.
- Разгибательные подошвенные ответы.
- Дизартрия (может начаться> 5 лет после начала).
- Арефлексия.
- Пирамидная слабость в нижних конечностях.
- Потеря положения суставов и чувствительности к вибрации в нижних конечностях.
- Потеря слуха и нарушение зрения (атрофия зрительного нерва или нистагм) являются обычными явлениями.

Другие клинические признаки или осложнения
[2, 3]- Ортопедия — сколиоз и симметричная полая стопа развиваются у большинства пациентов.
- Гипертрофическая кардиомиопатия — гипертрофия левого желудочка развивается примерно у 75% пациентов, а нарушения проводимости — у 10%. [2] Однако сердечные симптомы относительно редки и возникают на поздних стадиях заболевания. [4]
- Атрофия зрительного нерва встречается у 25-50% пациентов.
- Сахарный диабет развивается примерно у 10% пациентов. [9]
- Потеря слуха (обычно на поздних стадиях заболевания).
- Периферический цианоз, отеки и холодные ноги могут быть проблемой из-за снижения мышечной активности. [4]
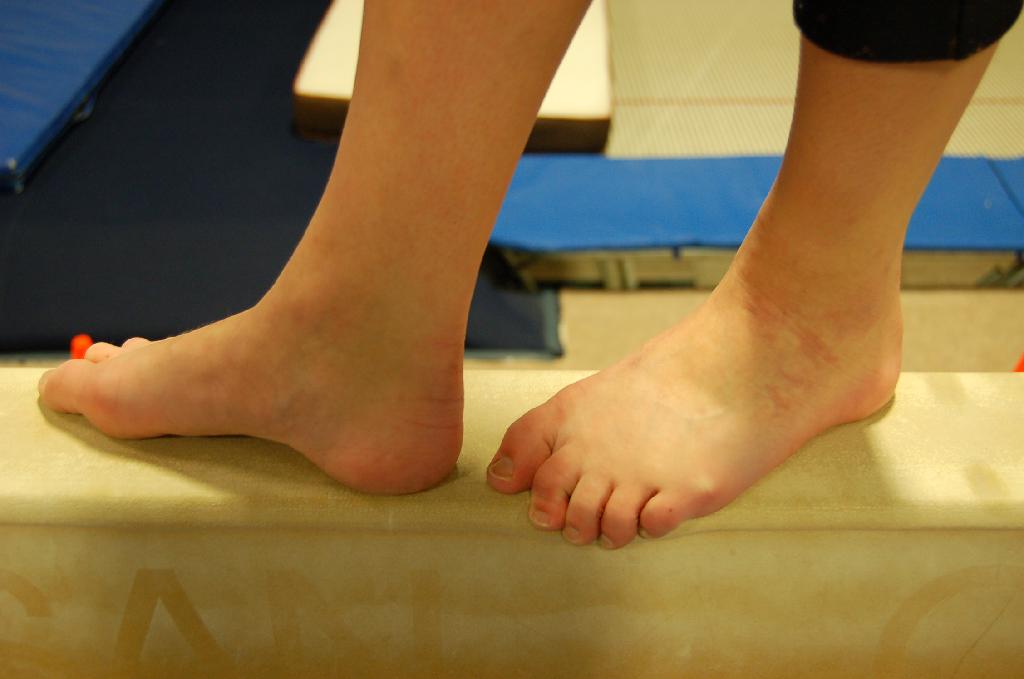
Дифференциальный диагноз
[2]- Дефицит витамина E может дать идентичную картину — важно диагностировать, поскольку она поддается лечению. [4]
- Дифференциальный диагноз широк, потому что многие заболевания могут вызывать раннее начало прогрессирующей атаксии с хроническим течением.
- Клинический анамнез и исследования должны быть направлены на выявление токсических, метаболических и иммунных заболеваний, паранеопластических заболеваний, пороков развития, опухолей задней черепной ямки, рассеянного склероза и лейкодистрофий.
- Атаксию Фридрейха нельзя с уверенностью исключить на основании клинических данных. Однако атаксия Фридрейха очень маловероятна при наличии ранней заметной атрофии мозжечка, общей неспособности к обучению и сохраненных потенциалов действия сенсорных нервов.
- Рассмотрите другие причины сколиоза или кардиомиопатии (если они проявляются симптомами).
Доступен алгоритм диагностики хронической прогрессирующей атаксии в детстве. [2]
Исследование и диагностика
[4]Первоначальные исследования
- Исследования нервной проводимости показывают, что скорость движения в руках более 40 мс-1 и отсутствие сенсорных потенциалов действия.
- Генетический анализ.
- ЭКГ — возможна гипертрофия желудочков и инверсия зубца T.
- Исключить дефицит витамина Е.
Дальнейшие исследования
[2]- Анализы крови: общий анализ крови, U&E и глюкоза
- Эхокардиография — может выявить гипертрофию желудочков, гипертрофию перегородки и гипертрофическую кардиомиопатию.
- МРТ головного и спинного мозга — показывает характерные атрофические изменения, особенно шейного отдела спинного мозга.
- Слуховые вызванные реакции ствола мозга и вызванные зрительные потенциалы — могут быть ненормальными.
- Генетическое консультирование и тесты — доступна пренатальная диагностика. Также возможно тестирование на носительство родственников пострадавших пациентов и их партнеров.
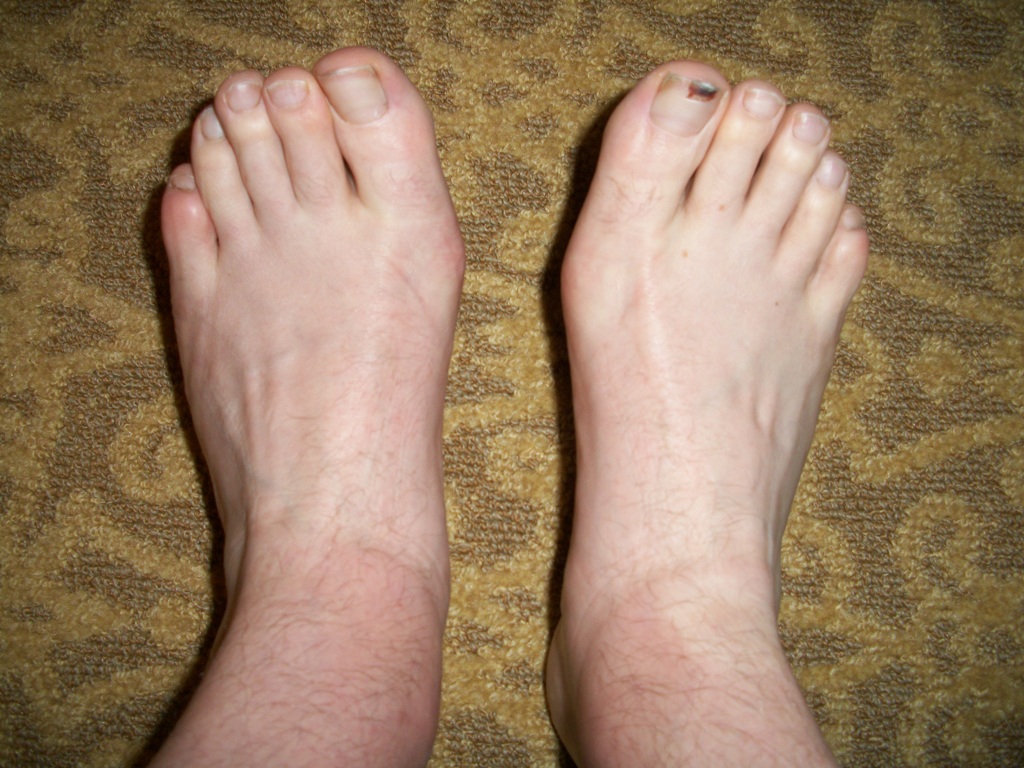
Менеджмент
[2, 4]Необходим междисциплинарный подход. Сюда должны входить невролог, генетик, генетический консультант, физиотерапевты, логопеды, терапевты по трудотерапии и социальные работники. Для решения конкретных проблем могут быть привлечены другие специальности — например, кардиологи, ортопеды и специалисты по диабету.
Атаксия Фридрейха — это мультисистемное заболевание. Ежегодные обзоры должны включать оценку неврологии, функцию сердца, опорно-двигательный аппарат, обзор комплексных систем и анализы крови (гематологию и контроль за диабет).
Возрастает интерес к разработке новых методов лечения, таких как клеточная и / или генная терапия. [10]
Специфическое медикаментозное лечение
До сих пор большинство исследований было сосредоточено на антиоксидантной терапии.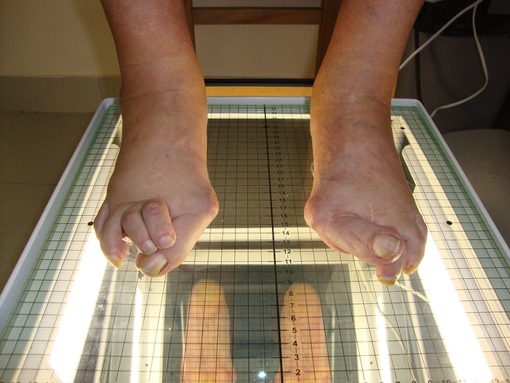
- Ни одно случайное контролируемое исследование с использованием идебенона или любого другого фармакологического лечения не показало значительного улучшения неврологических симптомов, связанных с атаксией Фридрейха. [11, 12]
- Другие возможные методы лечения включают деферипрон (хелатор железа), эритропоэтин, пиоглитазон и ингибиторы гистондеацетилазы (HDAC) и агонисты рецепторов, активируемых пролифератором пероксисом (PPAR), и гамма-агонисты. [13, 14]
Поддерживающая терапия
[2, 4]Может включать:
- Физиотерапию.
- Речевая и языковая терапия.
- Трудотерапия.
- Связь с образовательными и социальными службами.
- Лечение сердечной недостаточности и / или аритмий — стандартные методы лечения.
- Ортопедическая операция при наличии неприятных симптомов сколиоза, полой стопы или эквиноварусной деформации стоп.
- При деформации стопы также может помочь ботулотоксин или шины.
- Пассивные упражнения и разогревание при периферическом цианозе и холодных ногах.
- Диабет (если присутствует) обычно требует инсулина.
- Следует контролировать симптомы дисфункции сфинктера (например, срочность). Могут быть полезны уродинамическая оценка и лечение оксибутинином.
- При сексуальной дисфункции может потребоваться симптоматическое лечение.
- Консультации или антидепрессанты при депрессии (селективные ингибиторы обратного захвата серотонина (СИОЗС), вероятно, являются наиболее подходящими антидепрессантами).
- Дисфагия может потребовать изменения диеты или (на более поздних стадиях заболевания) гастростомии.
- Анестезия — пропофол, вероятно, следует избегать.
- Беременность — потенциальные осложнения — сердечная и респираторная недостаточность, тромбоэмболия легочной артерии и преждевременные роды. Сообщалось о необычной чувствительности к сульфату магния (при преэклампсии).
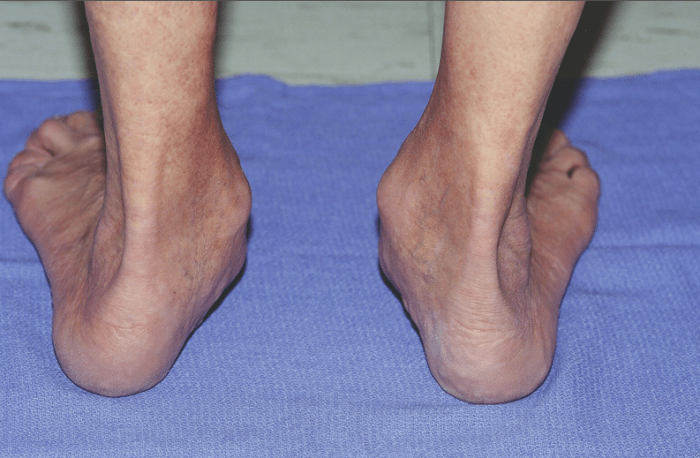
Прогноз
[2]- Средняя продолжительность жизни составляет 40-50 лет, но тяжесть заболевания и прогрессирование различаются; некоторые пациенты доживают до семидесяти лет.