Болезнь Хантингтона | Симптомы | Диагностика | Лечение
Болезнь Хантингтона (болезь Гентингтона, болезнь Хорея Гентингтона) – заболевание нервной системы, характеризующееся появлением периодических мышечных подергиваний или спазмов. Заболевание может развиться в любом возрасте, но чем раньше проявляются первые симптомы, тем стремительнее прогрессирует болезнь.
ПричиныЗаболевание возникает в результате постепенной дегенерации и отмирания нервных клеток и клеток головного мозга, что вызывается белком хантингтином. Причиной болезни Гентингтона становится наследственный фактор.
Симптомы болезни ХантингтонаХарактерные для болезни Хантингтона симптомы выглядят следующим образом:
Заболевание на первых стадиях протекает бессимптомно, первые признаки начинают проявляться в возрасте 35-40 лет.
Если Вы обнаружили у себя схожие симптомы, незамедлительно обратитесь к врачу. Легче предупредить болезнь, чем бороться с последствиями.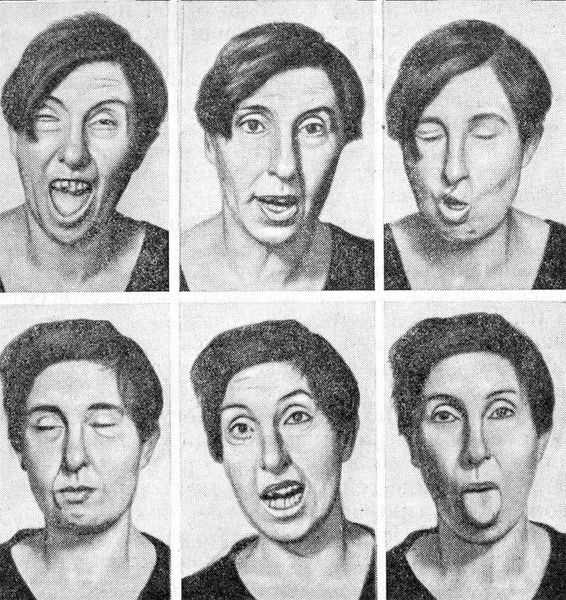
Чтобы определить, как лечить болезнь Хорея Гентингтона, врач — невролог проводит ряд исследований и анализов, среди которых:
Лечение болезни ХантингтонаНеобходимое для болезни Гентингтона лечение направлено на уменьшение симптомов и включает в себя:
- нейролептические препараты;
- бензодиазепины;
- противопаркинсонические средства;
- вальпроевую кислоту;
- психотропные препараты;
- транквилизаторы
В настоящее время болезнь Хантингтона неизлечима.
ОпасностьЕсли своевременно не заметить характерные для болезни Гентингтона симптомы и не начать лечение, это может привести к развитию осложнений:
- слабоумие;
- пневмония;
- ослабление иммунной системы;
- удушье;
- обезвоживание организма;
- сердечнососудистая недостаточность;
- повышенный риск травматизма.


Возможны попытки больного совершить суицид. Через 15-20 лет с момента появления первых признаков наступает летальный исход.
В группе риска находятся люди с генетической предрасположенностью (вероятность появления заболевания у детей больного человека составляет 50%).
ПрофилактикаДля профилактики заболевания беременным женщинам рекомендуется проходить перинатальную диагностику.
Данная статья размещена исключительно в познавательных целях и не является научным материалом или профессиональным медицинским советом.
сдать анализ в лаборатории KDLmed
Хорея Гентингтона — наследственное заболевание нервной системы, при котором происходит постепенная гибель определенных нервных клеток в головном мозге. При этом в первую очередь возникают двигательные нарушения. В более поздних стадиях болезни могут развиваться психические расстройства и снижение мыслительных функций.
Хорея Гентингтона встречается с частотой 4-8 случаев на 100 000 населения.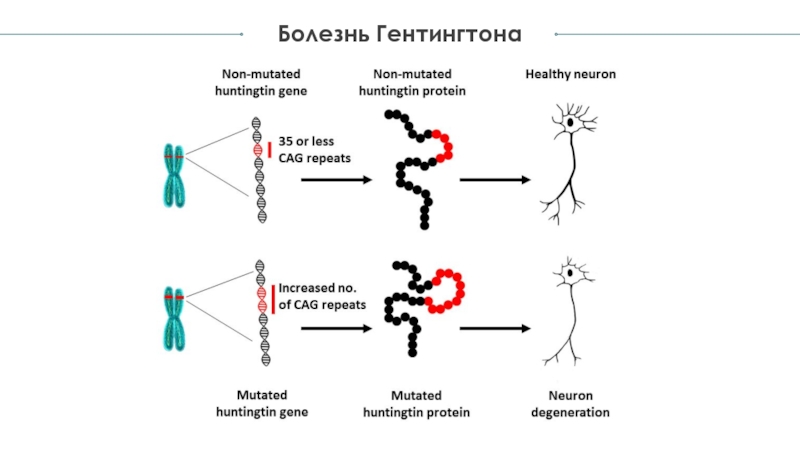 Первые признаки заболевания могут быть выявлены в 40 — 50 лет. С течением времени заболевание медленно развивается, вызывая гибель пациентов.
Первые признаки заболевания могут быть выявлены в 40 — 50 лет. С течением времени заболевание медленно развивается, вызывая гибель пациентов.
Хорея Гентингтона может начинаться в более раннем возрасте. При ювенильной форме болезни, симптомы возникают в возрасте до 20 лет. В таких случаях хорея Гентингтона имеет тяжелое течение и быстро прогрессирует.
Причиной заболевания является наследование дефектного гена, который кодирует строение определенного белка в нервных клетках. При этом происходит синтез данного белка с измененной структурой. Он начинает оказывать токсическое действие на определенные нервные клетки в головном мозге, в результате чего развивается гибель этих клеток. В основном страдают отделы головного мозга, ответственные за осуществление целенаправленных движений. Это приводит к возникновению неконтролируемых, беспорядочных движений (хореи) и других проявлений заболевания.
Хорея Гентигтона неизлечима. Лечение заключается в приеме препаратов, которые снижают выраженность некоторых симптомов.
Симптомы
Симптомы хореи Гентигтона:
- непроизвольные, неконтролируемые, внезапные движения (хорея)
- не координированные движения при выполнении каких-либо действий (например, жестикуляция, гримасничанье во время ходьбы)
- снижение мышечного тонуса (постоянного нормального напряжения мышц)
- ригидность (напряжение) мышц, общая скованность и замедленность движений (может развиваться при развитии некоторых черт болезни Паркинсона)
- медленные или скачкообразные движения глаз
- нарушение координации
- нарушение походки
- трудности при произношении слов
- нарушение глотания
- слюнотечение
Симптоматика умственных и психических расстройств при хорее Гентигтона:
- сложности в восприятии новой информации
- медлительность в выражении каких-либо мыслей
- трудности в планировании действий
- потеря способности контролировать свое поведение
- изменения в поведении (раздражительность, апатия, беспокойство)
- бессонница или чрезмерная сонливость
- мысли о смерти или суициде
- потеря интереса к окружающим событиям
- слабость, быстрая утомляемость
Общая информация
Хорея Гентингтона – наследственное заболевание, при котором происходит гибель нервных клеток в определенных структурах головного мозга, в результате чего развиваются двигательные нарушения, умственные и психические расстройства.
В норме в нервных клетках присутствует белок, который носит название хантингтин. Он связан со многими внутриклеточными компонентами, но роль данного белка в нервных клетках не известна.
Синтез хантигтина кодируется определенным геном. При возникновении дефекта данного гена происходит образование измененного белка (мутантного хантингтина). Данный белок нарушает нормальную деятельность нервных клеток, что приводит к их гибели.
При хорее Гентингтона в большей степени страдают структуры головного мозга, которые координируют движения человека. Повреждение данных структур приводит к развитию симптоматики заболевания.
Хорея Гентингтона имеет аутосомно-доминантный тип наследования. Это означает, что для возникновения данного заболевания у ребенка, только один родитель может иметь ген данного заболевания. Каждый родитель имеет две копии каждой хромосомы (содержат генетический материал). Ребенку передается по одному набору хромосом от каждого родителя.
В ситуациях, когда происходит передача гена заболевания по наследству, рано или поздно развиваются симптомы заболевания. Если наследования мутантного гена не произошло, то болезнь не развивается и будущему потомству не передается.
После начала заболевания функциональные способности пациента постепенно снижаются. Продолжительность жизни при хорее Гентингтона может составлять от 10 до 30 лет.
Развитие заболевания приводит пациентов к потере способности к самообслуживанию. На поздних стадиях болезни данные люди прикованы к кровати и требуют постоянного постороннего ухода.
У людей с хореей Гентингтона возникает склонность к суициду. Особенно высока вероятность суицидальных попыток после диагностики заболевания и в период снижения способности человека к самообслуживанию.
Наиболее частыми причинами смерти являются:
- пневмония – воспаление легких
- повреждения, полученные при падениях
- осложнения, связанные с нарушением глотания (например, пища может попадать в дыхательные пути, вызывая воспаление легких)
Кто в группе риска
К группе риска относятся:
- лица, имеющие близких родственников, страдающих данным заболеванием
- лица среднего возраста – проявления заболевания чаще возникают в данной возрастной группе.

Диагностика
Для диагностики заболевания проводится неврологический осмотр пациента, выявляются характерные признаки заболевания. Для подтверждения диагноза может быть проведено генетическое обследование, направленное на выявление мутантного гена хореи Гентингтона.
Проводятся также исследования головного мозга, с помощью которых удается выявить изменения в определенных отделах мозга.
Лабораторная диагностика:
- Генетический анализ. Для данного исследования берется кровь пациента. Затем производится анализ генетического материала (ДНК) для выявления специфической для хореи Гентингтона мутации в определенном гене.
- Определение наличия мутантного гена у эмбриона при экстракорпоральном оплодотворении.

Хорея Гентингтона — ДНК-диагностика хореи Гентингтона
Хорея Гентингтона (ХГ) – наследственное дегенеративное заболевание нервной системы, характеризующееся распространенными хореическими гиперкинезами (внезапными быстрыми, неритмичными непроизвольными движениями, возникающими беспорядочно в различных частях тела и усиливающимися при попытке совершить целенаправленное действие), деменцией (слабоумием) и имеющее неуклонно прогрессирующее течение. Клинические признаки
 Постепенно развиваются изменения психики (депрессии, тревога, раздражительность, эмоциональная неустойчивость, апатия, суицидальные попытки, разнообразные психозы) и деменция (нарушение памяти, замедление познавательных функций, снижение критического отношения к своему состоянию). Одной из наиболее ярких клинико-генетических особенностей болезни Гентингтона является феномен антиципации, т.е. нарастание тяжести болезни и появление ее в более молодом возрасте в последующих поколениях.
Постепенно развиваются изменения психики (депрессии, тревога, раздражительность, эмоциональная неустойчивость, апатия, суицидальные попытки, разнообразные психозы) и деменция (нарушение памяти, замедление познавательных функций, снижение критического отношения к своему состоянию). Одной из наиболее ярких клинико-генетических особенностей болезни Гентингтона является феномен антиципации, т.е. нарастание тяжести болезни и появление ее в более молодом возрасте в последующих поколениях.Тип наследования хореи Гентингтона- аутосомно-доминантный с полной пенетрантностью. Это означает, что половина детей больного (независимо от их пола) унаследуют от него мутантный ген и заболеют по достижении возраста дебюта заболевания.
Частота встречаемости: в среднем 5-10 случаев на 100 000 населения.
Ген гентингтина — HTT (IT15), повреждение которого приводит к болезни Гентингтона, картирован в локусе 4р16.3. В 5’-области этого гена содержится нестабильная последовательность тринуклеотидных повторов (CAG).
 В норме регистрируется от 11 до 35 CAG-повторов, а у больных хореей Гентингтона на одной из хромосом присутствует от 36 до 87 CAG-повторов. Большему числу CAG-повторов мутантного аллеля соответствует более ранний возраст начала заболевания, а также более быстрый темп его прогрессирования. Мутантные аллели нестабильны, что обычно приводит к нарастанию степени экспансии (увеличению числа повторов) в последующих поколениях. Мейотическая нестабильность мутантных аллелей значительно выше при передаче гена болезни по отцовской линии («эффект отцовской передачи»).
В норме регистрируется от 11 до 35 CAG-повторов, а у больных хореей Гентингтона на одной из хромосом присутствует от 36 до 87 CAG-повторов. Большему числу CAG-повторов мутантного аллеля соответствует более ранний возраст начала заболевания, а также более быстрый темп его прогрессирования. Мутантные аллели нестабильны, что обычно приводит к нарастанию степени экспансии (увеличению числа повторов) в последующих поколениях. Мейотическая нестабильность мутантных аллелей значительно выше при передаче гена болезни по отцовской линии («эффект отцовской передачи»).В Центре Молекулярной Генетики проводится прямая молекулярно-генетическая диагностика хореи Гентингтона, которая основана на определении числа CAG-повторов в гене HTT. Возможно проведение пресимптоматической, подтверждающей и дородовой диагностики в семьях, где наблюдаются случаи данного заболевания. Пресимптоматическая диагностика позоляет выявить носителей мутантного гена до появления у них клинический признаков заболевания (в молодом возрасте).
 Подтверждающая диагностика направлена на верификацию сомнительных клинических диагнозов наиболее точными молекулярно-генетическими методами. Дородовая (пренатальная) диагностика проводится на сроке от 8 недель беременности и позволяет устновить, унаследовал ли плод от больного родителя ген с экспансией CAG повтора. Для получения наиболее достоверных результатов необходимо предоставить биологический материал больного члена семьи.
Подтверждающая диагностика направлена на верификацию сомнительных клинических диагнозов наиболее точными молекулярно-генетическими методами. Дородовая (пренатальная) диагностика проводится на сроке от 8 недель беременности и позволяет устновить, унаследовал ли плод от больного родителя ген с экспансией CAG повтора. Для получения наиболее достоверных результатов необходимо предоставить биологический материал больного члена семьи.Нами разработан набор для регистрации экспансии CAG повтора в гене HTT методом ПЦР. Набор предназначен для использования в диагностических лабораториях молекулярно-генетического профиля.
Правила забора биологического материала для ДНК-анализа
При проведении пренатальной (дородовой) ДНК-диагностики в отношении конкретного заболевания, имеет смысл на уже имеющемся плодном материале провести диагностику частых анеуплоидий (синдромы Дауна, Эдвардса, Шерешевского-Тернера и др), пункт 54.1. Актуальность данного исследования обусловлена высокой суммарной частотой анеуплоидий — около 1 на 300 новорожденных, и отсутствием необходимости повторного забора плодного материала.
Публикации по теме раздела
Хорея ГентингтонаЛечение хорея гентингтона. Список клиник, рейтинг, отзывы, цены
О заболевании
Хорея Гентингтона или болезнь Гентингона является врожденной патологией головного мозга, которая поражает нервные клетки, а вследствие этого поражаются и определенные участки головного мозга. Когда человек болеет хореей Гентингтона, его поведение и когнитивные способности полностью меняются, в результате чего больной не может нормально думать и говорить. Со временем способность человека ходить и выражать свои мысли также нарушается. По данным Американского общества по изучению болезни Гентингтона, только в США этим недугом страдает 1 из 10 000 человек. В целом, в США этим заболеванием страдает 30 000 людей, а 150 000 -подвержены 50% риску развития патологии из-за генетической предрасположенности. Как правило, впервые заболевание проявляется в возрасте старше 35 лет. Иногда первые признаки патологии дают о себе знать в возрасте 45-50 лет. Доказано, что все случаи хореи Гентингтона вызваны генетическим нарушением, при котором патологический ген сверхэкспрессирует излишнее количество гентингтина. Этот белок отвечает за регуляцию моторики, поэтому первым симптомом хореи Гентингтона является странная неуклюжесть и нарушение равновесия. Со временем также поражаются другие белки головного мозга, следовательно, и другие участки головного мозга.
Доказано, что все случаи хореи Гентингтона вызваны генетическим нарушением, при котором патологический ген сверхэкспрессирует излишнее количество гентингтина. Этот белок отвечает за регуляцию моторики, поэтому первым симптомом хореи Гентингтона является странная неуклюжесть и нарушение равновесия. Со временем также поражаются другие белки головного мозга, следовательно, и другие участки головного мозга.
Симптомы
- Непроизвольные прерывистые движения
- Депрессия
- Резкие перепады настроения
- Запинание
- Невнятная речь
- Дезориентация
Диагностика
- Генетический анализ является очень важным для диагностирования хореи Гентингтона, так как он показывает, есть ли у человека дефектный ген, поражающий головной мозг. Современная медицина позволяет выполнять генетический анализ даже во время беременности (после 11 недели), если есть опасность, что мама или отец могут передать этот ген ребенку.
- КТ и МРТ воспроизводят трехмерные снимки головного мозга, которые показывают наличие повреждений головного мозга, если они присутствуют.

Виды лечения
- Консервативное лечение может только облегчить симптомы и замедлить прогрессирование заболевания. К сожалению, на сегодняшний день нет никаких методов лечения, которые бы могли полностью излечить хорею Гентингтона, например, препарат под названием Ксеназин помогает улучшить лишь двигательные способности человека. На данный момент в самых современных лабораториях также проводится генетический анализ, а ученые активно занимаются разработкой методов лечения хореи Гентингтона.
Автор: Доктор Вадим Жилюк
Прогрессирующая болезнь Хантингтона укоротила теломеры пациентов
US Department of Energy / Flickr
Ученые нашли еще одну болезнь, под действием которой теломеры становятся короче — это хорея Хантингтона.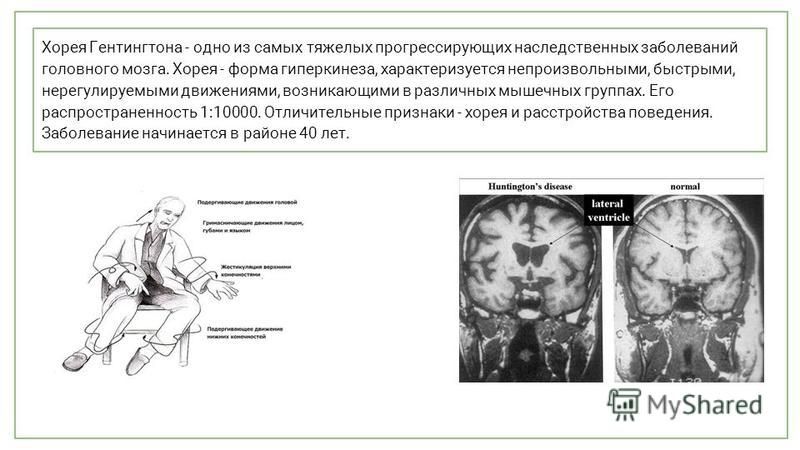 При этом существенные различия в длине теломер есть только тогда, когда болезнь уже проявилась и прогрессирует. А значит, теломеры — и, видимо, продолжительность жизни пациентов — сокращает не сам факт наличия мутации, а прогрессирующие нарушения в работе клеток. Работа опубликована в журнале Mechanisms of Ageing and Development.
При этом существенные различия в длине теломер есть только тогда, когда болезнь уже проявилась и прогрессирует. А значит, теломеры — и, видимо, продолжительность жизни пациентов — сокращает не сам факт наличия мутации, а прогрессирующие нарушения в работе клеток. Работа опубликована в журнале Mechanisms of Ageing and Development.
Длину теломер часто используют как маркер биологического возраста и как способ предсказать вероятность смерти для пациента. Ее проще измерить, чем большинство других маркеров, однако с ней возникает немало трудностей. Разброс по этому показателю довольно велик еще при рождении ребенка, а затем в течении жизни теломеры в отдельных клетках могут как укорачиваться, так и удлиняться. Поэтому ученые уже неоднократно приходили к заключению, что маркером должна служить не длина теломер сама по себе, а скорость ее изменения.
Несмотря на это, продолжают появляться работы, авторы которых измеряют длину теломер у людей в разных состояниях. Так, например, группа ученых под руководством Альберто Идальго-Браво (Alberto Hidalgo-Bravo) из Национального института реабилитации в Мехико собрала статистику по длине теломер у пациентов с болезнью Хантингтона.
Это заболевание вызвано необычной мутацией — накоплением повторов в гене митохондриального белка хантигтина. Чем больше повторов, тем белок работает хуже, но легче образует агрегаты и нарушает работу митохондрий. Исследователи отобрали 106 здоровых человек в качестве контроля (менее 36 повторов в гене), а также 100 человек с положительным результатом теста (более 36 повторов), из которых у 71 уже были клинические проявления болезни, а у 29 симптомы еще не возникли.
Авторы работы обнаружили, что у здоровых людей и у бессимптомных носителей мутации длина теломер существенно не отличалась (p = 0,597), а вот у пациентов с прогрессирующим заболеванием теломеры были в среднем в полтора раза короче (p = 0.011).
Что характерно, эти различия никак не были связаны с возрастом. Несмотря на то, что пациенты с симптомами болезни были в среднем несколько старше, чем в люди из двух других групп, ученые не нашли внутри ни одной из групп четкой зависимости длины теломер от возраста. Поэтому они заключили, что за укорочение теломер ответственны изменения, которые происходят в клетках при развитии болезни, а не сам факт наличия мутации.
Поэтому они заключили, что за укорочение теломер ответственны изменения, которые происходят в клетках при развитии болезни, а не сам факт наличия мутации.
Ученые предполагают, что механизм, который связывает мутантный белок с длиной теломер, выглядит следующим образом. Хантингтин накапливается в митохондриях и мешает им работать полноценно. В результате в них возникает больше активных форм кислорода, и в клетке развивается окислительный стресс. Активные формы кислорода могут выходить из митохондрии и повреждать теломеры (мы недавно рассказывали о том, что они особенно сильно подвержены действию стресса). Но мутантный белок обладает способностью блокировать белки репарации (мы писали и о том, как мутации в митохондриях мешают чинить мутации в ядерной ДНК), поэтому теломеры ломаются и становятся короче.
Против болезни Хантингтона до сих пор не существует эффективного лечения, но ученые ищут способы с ней справиться — например, с помощью аутофагии или антисмысловых цепочек нуклеотидов.
Полина Лосева
Хорея Гентингтона (хореическая деменция) | МКДЦ ФГБНУ НЦН
Хорея – гиперкинез, характеризующийся беспорядочными разбросанными подергиваниями мышц конечностей (чаще верхних), туловища и лица.
КЛИНИЧЕСКАЯ КАРТИНА
Больные с такими гиперкинезами непоседливы, суетливы, постоянно гримасничают. В результате этого часто ушибаются об окружающие предметы, с большим трудом и довольно недолго выдерживают заданную позу. Подергивания прерываются произвольными движениями, которые выглядят некоординированно. Для больных характерна танцующая походка. Возникает дизартрия вследствие оральных гиперкинезов, в тяжелых случаях больные совсем не говорят. В развитии хореи играет роль поражение стриарных структур и мозжечка. В зависимости от причин возникновения различают хорею беременных, хорею Сиденгама (малую), хорею Гентингтона.
Хорея беременных – вариант малой хореи. Возникает в первые месяцы беременности у молодых женщин. Чаще в анамнезе у них имеются данные о перенесенной в детстве хорее.
Малая хорея Сиденгама – результат ревматического поражения мозга. Возникает чаще у детей. Происходят воспалительные, сосудистые и дегенеративные поражения нервной ткани. Характерны некоординированные, непроизвольные движения, значительное снижение мышечного тонуса, эмоциональная лабильность, агрессивность, слезливость, импульсивность. Характерен симптом Гордона (тоническая форма коленного рефлекса).
Хорея Гентингтона – это наследственное заболевание, возникающее в период с 30 до 40 лет. Сначала проявляются хореические гиперкинезы, позже – прогрессирующее слабоумие, полный распад личности.
Дифференциальную диагностику следует проводить с другими хореями, старческой хореей. Решающее значение имеет семейный анамнез.
ЛЕЧЕНИЕ
Лечение должно производиться исключительно врачом-неврологом. Самолечение недопустимо. Лечение гиперкинезов и импульсивности проводят нейролептиками. Прогноз заболевания неблагоприятный. Больные умирают на фоне кахексии, глубокого слабоумия. Лицам с отягощенной наследственностью по данному заболеванию не рекомендуется иметь детей.
Болезнь Гентингтона у ребенка. Первое описание заболевания у ребенка в Украине. Собственное наблюдение
Modern pediatrics. Ukraine. 2019.4(100):60-65; doi 10.15574/SP.2019.100.60
Стеценко Т. И., Савченко Е. И., Салан Н. З., Головатюк И. В.
Национальная медицинская академия последипломного образования имени П.Л. Шупика, г. Киев, Украина
Национальная детская специализированная больница «ОХМАТДЕТ» МЗ Украины, г. Киев
Для цитирования: Стеценко ТИ, Савченко ЕИ, Салан НЗ, Головатюк ИВ. (2019). Болезнь Гентингтона у ребенка. Первое описание заболевания у ребенка в Украине. Собственное наблюдение Современная педиатрия. Украина. 4(100):60-65; doi 10.15574/SP.2019.100.60
Статья поступила в редакцию 18.02.2019 р., принята в печать 19.05.2019 р.
Болезнь Гентингтона (ХГ, МКХ-10 — G-10, МКХ-11 — 8A01.10) — аутосомно-доминантное нейродегенеративное заболевание, которое характеризуется двигательными нарушениями (хорея у взрослых и акинетико-ригидный синдром у детей), психическими расстройствами и деменцией c прогрессирующим течением и летальностью 100%.
Цель: представить уникальный случай диагностированного заболевания хореи Гентингтона у ребенка в возрасте 4 лет в третьем поколении в семье. В Украине данный случай описывается впервые.
Результаты: Представляем собственный опыт наблюдения и лечения девочки 4 лет 7 месяцев, которая поступила в НДСБ «ОХМАТДЕТ» с жалобами на потерю разговорных и психических навыков, нарушение походки и наличие приступов. С 4-летнего возраста начался регресс психических функций (пропал интерес к сказкам, стала невнимательной, ухудшилась память, перестала себе обслуживать) и речи с формированием экстрапирамидной дизартрии и обеднением словарного запаса. В это же время ухудшилась походка, появились генерализированные эпилептические приступы. Необходимо акцентировать внимание на том, что семейный анамнез отягощен по линии отца — у отца, деда, прабабушки по отцовской линии хорея Гентингтона. При осмотре у ребенка были выявлены нарушения коммуникации, дисфазия, экстрапирамидная дизартрия, псевдобульбарный синдром; походка — спастико-дистоническая, хуже слева; изменение мышечного тонуса по пластическому типу в руках и ногах, больше слева; гиперкинезы в пальцах рук (атетоз), дистония в стопах, клонусы стоп. На МРТ отмечались умеренно выраженные диффузное уменьшение объема паренхимы обоих полушарий мозжечка, углубленная и расширенная борозда мозжечка, некоторое утончение мозжечковых ножек; также отсутствовали миндалины мозжечка, широкое отверстие Мажанди и умеренно увеличенная в объеме большая цистерна. На электроэнцефалограмме была выявлена генерализированная высокоамплитудная пик-волновая эпилептическая активность во время всей записи. В возрасте 4 лет был повторно проведен молекулярно-генетический анализ, количество ЦАГ-повторов составило 130 и был подтвержден диагноз болезни Гентингтона.
Выводы. 1. Для болезни характерен генетический импринтинг — зависимость формы и тяжести проявлений заболевания от отцовского источника мутантной хромосомы. 2. В детском и юношеском возрасте доминирует акинетико-ригидный синдром. 3. В детском возрасте (до 10 лет) болезнь может дебютировать эпилептическими приступами, поэтому необходимо собирать семейный анамнез не только по эпилепсии, но и относительно неврологических и психиатрических заболеваний, в тому числе у взрослых. 4. При признаках прогрессирующего нейродегенеративного заболевания у ребенка не следует отбрасывать возможность диагноза хореи Гентингтона нужно направлять такие семьи на медико-генетическое консультирование.
Исследование было выполнено в соответствии с принципами Хельсинской Декларации. Протокол исследования был одобрен Локальным этическим комитетом (ЛЭК) всех участвующих учреждений. На проведение исследований было получено информированное согласие родителей ребенка.
Авторы заявляют об отсутствии конфликта интересов.
Ключевые слова: дети, нейродегенеративные болезни, эпилепсия, хорея.
ЛИТЕРАТУРА
1. Aicardi J, Bax M., Gillberg Ch. (2013). Diseases of the nervous system in children. 3th edition. Moscow. Binom: 383—386.
2. Barcovich AJ, Raubaud Ch. (2012). Padiatric neuroimaging. 5th edition. Philadelphia. Lippincott: 383—386.
3. Choudhary A, Minocha P, Sitaraman S. (2017). A case report of juvenile Huntington disease. Journal of Pediatric and Neonatal Individualized Medicine. 6(2): e060217. https://doi.org/10.5582/irdr.2016.01084; PMid:28357189 PMCid:PMC5359361
4. Fahn S. (2000). Huntington disease. In: Merrit`s Neurology 10th edition. Eds.: L.P. Rowland. Lippincott, Philadelphia: 659—662.
5. Kailash Chandra Patra, Mukund Sudhir Shirolkar. (2015). Childhood-on-set (Juvenile) Huntington’s disease: A rare case report. J Pediatr Neurosci. 10(3): 276—279. https://doi.org/10.4103/1817-1745.165709; PMid:26557176 PMCid:PMC4611904
6. Kendrick LM, Hudgell D, Hellman A, Weaver MS. (2019). Attending to Total Pain in Juvenile Huntington Disease: A Case Report Informed by Narrative Review of the Literature. J. Palliat Care. 5: 825859719835560. https://doi.org/10.1177/0825859719835560; PMid:30950323
7. Latimer CS, Flanagan ME, Cimino PJ, Jayadev S, Davis M, Hoffer ZS, Montine TJ, Gonzalez-Cuyar LF, Bird TD, Keene CD. (2017) Neuropathological Comparison of Adult Onset and Juvenile Huntington’s Disease with Cerebellar Atrophy: A Report of a Father and Son. J Huntingtons Dis. 6(4): 337—348. https://doi.org/10.3233/JHD-170261; PMid:29036832 PMCid:PMC5832043
8. Lyon G, Kolodny EH, Pastores GM. (2006). Neurology of Hereditary Metabolic Diseases of Children. 34-edition. NY: McGraw4Hill: 281—284.
9. Mukhin KYu, Oysyachina ID, Savvin DA, Piliya SV, Petrukhin AS. (2010). Childhood onset form of Huntingnon’s disease (clinical case). Russkiy zhurl detskoy nevrologii. 1(5): 35—42.
10. Shi-Shuang Cui, Ru4Jing Ren, Ying Wang, Gang Wang, Sheng-Di Chen. (2017). Tics as an initial manifestation of juvenile Huntington’s disease: case report and literature review. BMC Neurology. https://doi.org/10.1186/s12883-017-0923-1; PMid:28789621 PMCid:PMC5549341
11. Weinmann ARO, Souza GA, Souza GA, Sandim GB, Giuliani LR. (2017). Case Report: Huntington’s disease in four years old male. European journal of paediatric neurology. 2(1): 61—62. https://doi.org/10.1016/j.ejpn.2017.04.924
Текст статьи
Содержание
Хорея и болезнь Хантингтона
Предоставлено Shu-Leong Ho, MD, FRCP
Генри Г. Леонг, профессор и заведующий отделением Гонконгского университета,
Больница Королевы Марии
Отделение неврологии, медицинский факультет
Гонконг
Хорея
Обновления 2019 г. Автор:
Эстер Кубо, MD, PhD
Невролог
Больница Университарио Бургос
Бургос, Испания
Марк Гуттман, MD, FRCPC
Директор
Центр двигательных расстройств
Торонто, Онтарио, Канада
Брэндон Бартон, Мэриленд
Доцент
Медицинский центр Университета Раша
Чикаго, штат Иллинойс, США
Хорея — ненормальное непроизвольное движение, образованное от греческого слова «танец».Для него характерны короткие, резкие, нерегулярные, непредсказуемые, нестереотипные движения. В более легких случаях хорея может казаться целенаправленной. Пациент часто кажется суетливым и неуклюжим. В целом хорея может поражать различные части тела, мешать речи, глотанию, осанке и походке, а также исчезает во сне.
Диагностика является сложной задачей, поскольку хорея имеет одинаковую феноменологию независимо от ее этиологии. Хорею обычно классифицируют как первичную (идиопатическую, наследственную) или вторичную (приобретенную).Наследственные хореиформные расстройства имеют тенденцию к развитию незаметно и обычно симметричны, тогда как приобретенная хорея чаще бывает острой или подострой и может быть асимметричной или односторонней. Существует широкий спектр, казалось бы, несвязанных причин, от беременности (chorea gravidarum) до наследственных форм, таких как болезнь Хантингтона и доброкачественная наследственная хорея, инфекционных / иммунных, таких как хорея Сиденхама и системная красная волчанка, очаговых сосудистых поражений в базальных ганглиях, лекарственные средства, такие как леводопа, нейролептики и оральные контрацептивы, или различные метаболические и эндокринологические нарушения, такие как гипертиреоз, гипо / гиперпаратиреоз и гипо / гипергликемия.Его патофизиология включает функциональную дисрегуляцию моторной цепи базальных ганглиев, где конечный таламо-кортикальный выброс увеличивается, что приводит к увеличению движений и хореи. Нарушение схемы базальных ганглиев может быть вызвано структурным повреждением, избирательной дегенерацией нейронов, блокадой рецепторов нейромедиаторов, метаболическими нарушениями или аутоиммунными состояниями.
Лечение хореи заключается в устранении ее корневой этиологии. К сожалению, не было доказано, что ни один агент замедляет или останавливает прогрессирование наследственной хореи, за исключением медь-снижающих терапий при болезни Вильсона.Наиболее распространенные симптоматические методы лечения хореи включают использование средств, блокирующих дофаминовые рецепторы, и истощителей дофамина, таких как тетрабеназин, валбеназин и дейтетрабеназин. Синдромы аутоиммунной хореи, такие как системная красная волчанка (СКВ) и синдром антифосфолипидных антител, также могут поддаваться лечению глюкокортикоидами, плазмообменом или внутривенным иммуноглобулином (ВВИГ).
Нажмите здесь, чтобы посмотреть видео с изображением хореи (только для участников)
Болезнь Хантингтона
Предоставлено Hubert Fernandez, MD
Голова, двигательные расстройства
Кафедра неврологии
Клиника Кливленда
Кливленд, Огайо, США
Обновление 2019 г., Эстер Кубо, доктор медицинских наук, и Марк Гутман, доктор медицинских наук, FRCPC
Болезнь Хантингтона — аутосомно-доминантное нейродегенеративное заболевание (таким образом, вероятность развития болезни у каждого ребенка пораженного родителя составляет 50%).Это вызвано экспансией тринуклеотидного повтора цитозин-аденин-гуанина (CAG) в гене хантингтина (HTT) на хромосоме 4p. У большинства людей болезнь Гентингтона развивается в возрасте от 30 до 54 лет, но она может проявляться уже в возрасте от 4 до 80 лет. Распространенность HD во всем мире составляет примерно 2,7 на 100 000 человек.
Болезнь Хантингтона клинически характеризуется триадой моторных, когнитивных и психиатрических симптомов. Двигательные особенности включают: нарушение непроизвольных (хорея) и произвольных движений; снижение ловкости рук, невнятная речь, трудности с глотанием, проблемы с равновесием и падения.Он также может проявляться паркинсонизмом и дистонией (чаще встречается в молодом возрасте, так называемый Вестфальский вариант). Когнитивные особенности изначально характеризуются потерей скорости и гибкости мышления, но позже перерастают в глобальную деменцию. Психиатрические особенности могут включать: депрессию (наиболее частую), манию, обсессивно-компульсивное расстройство, раздражительность, беспокойство, возбуждение, импульсивность, апатию и социальную изоляцию.
Окончательный диагноз ставится на основании генетического тестирования и подтверждается результатами анализа целевых мутаций тринуклеотидной экспансии CAG из ≥36 повторов в гене HTT.Прогностическое генетическое тестирование — это вариант для людей, которые подвержены риску развития HD, категории, которая включает бессимптомных лиц с положительным семейным анамнезом HD или пациентов с положительным семейным анамнезом, у которых есть продромальные симптомы (например, раздражительность, беспокойство, депрессия, или когнитивное нарушение), которые предполагают приближающееся начало симптоматического HD. Перед проведением прогностического генетического тестирования рекомендуется предварительное генетическое консультирование. Для семей HD возможно доимплантационное генетическое тестирование и, следовательно, выбор эмбрионов для переноса без генетической мутации HD.Пренатальное тестирование проводится только в тех случаях, когда мать рассматривает возможность прерывания беременности в случае положительного результата теста плода. В остальном пренатальное тестирование — это то же самое, что и тестирование бессимптомного ребенка, что не рекомендуется с этической точки зрения.
Для пациента с хореей, нетипичного для HD или отрицательного для генетической мутации HD, дифференциальный диагноз широк и включает наследственные и приобретенные причины хореи. В этих случаях МРТ головного мозга может быть полезной для установления альтернативного диагноза, такого как ишемический инфаркт, нейродегенерация, связанная с пантотенаткиназой, рассеянный склероз, новообразование или болезнь Крейтцфельдта-Якоба.В противном случае следует указать дополнительное обследование на основании медицинского и семейного анамнеза, физического и неврологического обследования.
Наилучший уход за пациентами с HD обеспечивается многопрофильной командой медицинских специалистов и поддерживающих лиц, которые удовлетворяют широкие физические и психологические потребности пациентов и их семей и решают новые проблемы по мере их возникновения посредством долгосрочного наблюдения. Агенты, блокирующие дофаминовые рецепторы, или агенты, истощающие дофамин, могут использоваться, если хорея ухудшает качество жизни.Психиатрические симптомы, такие как депрессия, психоз и возбуждение, управляются так же, как и с любым другим психическим заболеванием. Важны нефармакологические подходы к некоторым функциональным трудностям и поведенческой дисфункции при болезни Хантингтона Немедикаментозные вмешательства включают логопедию и диетическое обслуживание для лечения дисфагии и потери веса, физиотерапевта для лечения нарушения походки и падений. для деменции, связанной с HD.Сообщалось о стереотаксических хирургических вмешательствах (DBS) в тяжелых случаях хореи, приводящих к инвалидности. В настоящее время ведутся исследования методов лечения, таких как подавление генов с помощью методов антисмысловых олигонуклеотидов.
Болезнь Хантингтона — лучший канал здоровья
Болезнь Хантингтона (также известная как болезнь Хантингтона) — это неврологическое заболевание (нервная система), вызванное наследованием измененного гена. Гибель клеток мозга в определенных областях мозга приводит к постепенной утрате когнитивных (мышление), физических и эмоциональных функций.Болезнь Хантингтона — сложное и изнурительное заболевание, от которого нет лекарства.Наиболее частый симптом — судорожные движения рук и ног, известные как «хорея». Хорея обычно начинается с легких подергиваний и постепенно усиливается с годами. Человек с болезнью Гентингтона также может иметь трудности с речью, глотанием и концентрацией внимания.
Генетика болезни Хантингтона
Болезнь Хантингтона вызывается измененным геном. Этот ген передается от родителей к ребенку, но при рождении это состояние не очевидно.Симптомы обычно, но не всегда, впервые появляются, когда человек приближается к среднему возрасту. Болезнь Хантингтона — это медленное, прогрессирующее заболевание, которое по-разному поражает людей.
Человек с болезнью Гентингтона может жить от 15 до 25 лет после появления первых симптомов. Диагноз ставится на основании семейного анамнеза болезни Хантингтона (если известно), генетического тестирования, а также оценки физических, неврологических и эмоциональных симптомов. Лекарства от болезни Хантингтона нет.
Тестирование генов на болезнь Гентингтона
Ребенок, рожденный от человека, несущего ген болезни Хантингтона, имеет 50-процентную вероятность унаследовать этот ген и заболеть.Люди из группы риска могут пройти тест, чтобы узнать, унаследовали ли они этот ген. Человек должен быть не моложе 18 лет и желать знать свой генный статус, прежде чем он сможет пройти тест. Решение о прохождении теста — личное дело каждого. Консультации доступны, чтобы помочь человеку с болезнью Хантингтона и его семье, опекунам и друзьям справиться с генетическим результатом.
Узнайте больше о генетических услугах и генетическом консультировании.
Симптомы болезни Гентингтона
Симптомы болезни Хантингтона делятся на три типа: физические, когнитивные и эмоциональные.
Физические симптомы включают:
- Легкое подергивание пальцев рук и ног
- Отсутствие координации и склонность к опрокидыванию предметов
- Трудности при ходьбе
- Танцевальные или судорожные движения рук или ног (хорея)
- Речь и трудности с глотанием.
- Кратковременная потеря памяти
- Трудности с концентрацией внимания и составлением планов.
- Депрессия (около трети людей с болезнью Гентингтона испытывают депрессию)
- Поведенческие проблемы
- Перепады настроения, апатия и агрессия.
Депрессия
Считается, что около трети людей с болезнью Гентингтона испытывают депрессию. Симптомы депрессии, такие как отсутствие влечения, должны быть исследованы с медицинской точки зрения, а не просто рассматриваться как часть процесса болезни.
Рекомендации для членов семьи, друзей и опекунов:
- Обратитесь к врачу для диагностики. Существуют лекарства для лечения депрессии.
- Психотерапия может быть вариантом.
- Регулярные упражнения и умеренное пребывание на солнце могут помочь облегчить депрессию.
- Постарайтесь включить в свой распорядок больше занятий, которые особенно нравятся этому человеку.
Болезнь Хантингтона и поведенческие проблемы
Поведенческие проблемы, связанные с болезнью Гентингтона, как полагают, вызваны сочетанием событий, включая повреждение мозга по мере прогрессирования болезни, а также понятные расстройства и депрессию, которые люди испытывают, когда сталкиваются с хроническими заболеваниями. болезнь.
Не все люди с болезнью Гентингтона будут испытывать одни и те же поведенческие проблемы, поскольку болезнь поражает людей по-разному.Серьезность поведенческих изменений может варьироваться от легкой и едва заметной до чрезвычайно разрушительной. Для семьи, друзей и опекунов важно понимать, что изменения в поведении человека являются частью болезни и не находятся под их сознательным контролем.
Потеря мотивации
Разделы мозга, которые помогают нам планировать, организовывать и начинать действия, поражены болезнью Хантингтона. Человек может показаться ленивым, потому что он ничего не будет делать (кроме, например, лежать в постели или смотреть телевизор), если предоставлен самому себе.
Рекомендации для членов семьи, друзей и опекунов:
- Поймите, что крик или споры не могут мотивировать человека.
- Человек может хорошо реагировать на действия с другими, поэтому возьмите на себя инициативу и побудите их следовать.
- Помощь человеку в участии повышает его чувство собственного достоинства, что жизненно важно для снижения риска депрессии.
Потеря последовательности задач
Задачи должны выполняться в определенном порядке.Например, для мытья посуды необходимо наполнить раковину горячей водой с моющим средством, вымыть посуду, высушить ее и убрать. Человек с болезнью Гентингтона может запоминать части задания, но не в правильном порядке. Они могут наполнить раковину горячей водой, но потом убрать грязные тарелки, не мыть их.
Рекомендации для членов семьи, друзей и опекунов:
- Наблюдать за человеком и помогать ему выполнять задания в надлежащей последовательности.
- Поощряйте привычку делать одно дело за раз.
Неспособность блокировать отвлекающие факторы
Принятие пищи во время просмотра телевизора или прослушивания музыки может быть очень трудным для человека с болезнью Хантингтона, поскольку он не может сосредоточиться на обоих вещах одновременно. Вот почему рекомендуется принимать пищу в спокойной обстановке.
Рекомендации для членов семьи, друзей и опекунов:
- Поощряйте привычку делать одно дело за раз.
- Помните, что занятия, которые мы принимаем как должное, например ходьба, могут потребовать концентрации от человека с HD.Возможно, они не смогут одновременно вести беседу.
Сниженные способности
Человек с болезнью Хантингтона может показаться более беспечным. Например, они могут не убирать дом должным образом или могут не соблюдать свои обычные стандарты личной гигиены.
Рекомендации для членов семьи, друзей и опекунов:
- Цените, что человек старается изо всех сил. На их работоспособность влияет болезнь, а не лень.
- Человек может даже не осознавать, что сделал ошибку. Возьмите за правило проверять от их имени.
- Постарайтесь установить строгий распорядок купания.
- Не отказывайте им в задачах просто потому, что их легче делать самостоятельно — продолжать вносить свой вклад очень важно для чувства собственного достоинства человека.
Неприемлемое социальное поведение
Осведомленность о социальных условностях может снизиться, что приведет к (например) непристойным или грубым комментариям в адрес других.У некоторых людей с болезнью Гентингтона больше нет «сдерживающих» эмоций стыда, смущения и страха, которые помогают контролировать социальное поведение.
Рекомендации для членов семьи, друзей и лиц, осуществляющих уход, включают:
- Объяснение несоответствия их поведения может быть им недоступно. Кричать и спорить, вероятно, не получится.
- Человек может не понимать неприемлемости своего поведения, но он может придерживаться правил, если вы их устанавливаете.
- Возможно, вам стоит подумать об ограничении ваших общественных мероприятий.
Раздражительность и агрессия
Некоторые люди с болезнью Гентингтона легко раздражаются или злятся. Частично это может быть вызвано неспособностью видеть вещи с точки зрения другого человека. Некоторые люди с болезнью Гентингтона могут казаться эгоистичными и эгоистичными.
Рекомендации для членов семьи, друзей и опекунов:
- Помните, что болезнь мешает человеку мыслить гибко. Они могут чувствовать себя более комфортно и непринужденно в знакомой обстановке и ситуациях.
- Убедитесь, что у них есть достаточный контроль над своими вариантами. Например, человек может стать нервным и раздражительным, если ему не разрешают выбирать себе одежду в течение дня.
- Посмотрите, что может представлять собой поведение. Например, человек может выплевывать пищу, потому что у него слишком много во рту, а не потому, что он намеренно пытается рассердить.
- Помните общее правило: по одному. Попытки сделать две вещи одновременно могут вызвать волнение.
- Сосредоточьтесь на позитивном поведении и постарайтесь как можно больше игнорировать остальное.
- Компромисс в любовных отношениях нарушается болезнью Хантингтона, так как человек, возможно, потерял решающие эмоции из-за болезни.
Трудности с общением
По мере прогрессирования болезни части мозга, которые помогают контролировать мышцы лица, горла и языка, поражаются все чаще. Это может вызвать у человека значительные проблемы с речью.Человек также может не начинать разговоры, так как ответственные за это участки мозга также повреждены.
Рекомендации для членов семьи, друзей и опекунов:
- Не думайте, что они не могут понять, о чем вы говорите, только потому, что не могут сформулировать ответ. Болезнь обычно не влияет на понимание.
- Не торопите их. Выделите достаточно времени для их ответа.
- Если вы видите, что человеку трудно выразить себя, спросите, не нужна ли ему помощь, вместо того, чтобы делать это автоматически.Например, человеку может не нравиться, что вы заканчиваете его слова или предложения без его разрешения.
- По возможности предлагайте им выбор вместо открытых вопросов. Например, «Хотите пасту или рыбу на ужин?» Легче ответить, чем «Что вы хотите съесть?».
- Флэш-карточки с типичными ответами, такими как «да» и «нет», могут быть полезны.
- Если человек прогрессирует до потери речи, продолжайте разговаривать с ним как обычно. В противном случае вы рискуете заставить их почувствовать себя изолированными и невидимыми.
Поддержка людей с болезнью Гентингтона
Индивидуальные работники и работники службы поддержки семей наняты во всех регионах для поддержки людей с болезнью Гентингтона, групп риска и их семей.
Услуги включают:
- Информация и консультации
- Обследование и направление
- Практическая помощь с поддержкой на дому
- Практическая помощь с размещением и передышкой
- Постоянная поддержка лиц, осуществляющих уход,
- Праздничные и волонтерские программы.
Куда обратиться за помощью
Болезнь Хантингтона: симптомы, причины и лечение
Болезнь Хантингтона — неизлечимое наследственное заболевание головного мозга, которое повреждает клетки мозга. Он оказывает разнообразное воздействие, влияя на движение, мышление и настроение.
Болезнь Хантингтона возникает, когда дефектный ген заставляет токсичные белки накапливаться в головном мозге.
Болезнь Хантингтона поражает 3–7 человек на каждые 100 000 человек европейского происхождения. По данным Genetics Home Reference, это реже встречается у людей японского, китайского и африканского происхождения.
Первые признаки обычно появляются в возрасте от 30 до 50 лет.
Болезнь Хантингтона — неврологическое заболевание. Это наследственное заболевание, которое возникает из-за дефектных генов. Токсичные белки накапливаются в головном мозге и вызывают повреждения, вызывающие неврологические симптомы.
Поскольку болезнь поражает разные части мозга, она влияет на движения, поведение и познание. Становится труднее ходить, думать, рассуждать, глотать и говорить. В конце концов, человеку потребуется постоянный уход.Осложнения обычно заканчиваются летальным исходом.
В настоящее время лекарства нет, но лечение может помочь с симптомами.
Признаки и симптомы чаще всего появляются в возрасте от 30 до 50 лет, но они могут появиться в любом возрасте. В конце концов болезнь или ее осложнения могут закончиться летальным исходом.
Ключевые симптомы включают:
Развитие симптомов может различаться у разных людей. Некоторые люди сначала испытывают депрессию, а затем изменяют моторику. Изменения настроения и необычное поведение — частые ранние признаки.
Ранние признаки и симптомы
Врач может не распознать ранние симптомы, если в семье не было ранее диагноза болезни Гентингтона. Для постановки диагноза может потребоваться время.
Первоначальные признаки и симптомы включают:
- небольшие неконтролируемые движения
- небольшие изменения в координации и неуклюжесть
- спотыкание
- легкие признаки настроения и эмоциональные изменения
- трудности с концентрацией внимания и функционированием в школе или на работе
- кратковременные упущения- термическая память
- депрессия
- раздражительность
Человек может потерять мотивацию и сосредоточенность.Они могут казаться вялыми и безынициативными.
Другие возможные признаки болезни Хантингтона могут включать спотыкание, падение вещей и забывание имен людей. Однако большинство людей делают это время от времени.
Средняя и поздняя стадии
Со временем симптомы становятся более серьезными.
К ним относятся физические изменения, потеря контроля над движениями, а также эмоциональные и когнитивные изменения.
Физические изменения
Человек может испытывать:
- трудности с речью, в том числе поиск слов и невнятную речь
- потеря веса, ведущая к слабости
- трудности с приемом пищи и глотанием, так как мышцы рта и диафрагмы могут не работать должным образом
- риск удушья, особенно на более поздних стадиях
- неконтролируемые движения
Могут быть неконтролируемые движения тела, в том числе:
- неконтролируемые движения лица
- подергивания частей лица и головы
- подергивания или суетливые движения рук, ног и тела
- покачивание и спотыкание
По мере прогрессирования болезни Хантингтона неконтролируемые движения возникают чаще и обычно с большей интенсивностью.В конце концов они могут стать медленнее, поскольку мышцы станут более жесткими.
Эмоциональные изменения
Они могут сменяться, а не происходить постоянно.
К ним относятся:
- агрессия
- гнев
- антисоциальное поведение
- апатия
- депрессия
- возбуждение
- разочарование
- отсутствие эмоций становится более очевидным
- капризность
- упрямство
- когнитивные изменения
Есть может быть:
- потеря инициативы
- потеря организационных навыков
- дезориентация
- трудности с фокусировкой
- проблемы с многозадачностью
Более поздняя стадия
В конце концов, человек больше не сможет ходить или говорить , и им потребуется полный уход.
Однако они обычно понимают большую часть того, что слышат, и будут знать о друзьях и членах семьи.
Неспособность делать то, что раньше было легким, может привести к разочарованию и депрессии.
Похудание может усугубить симптомы и ослабить иммунную систему пациента, делая их более уязвимыми для инфекций и других осложнений.
Болезнь Хантингтона сама по себе обычно не приводит к летальному исходу, но может иметь место удушье, пневмония или другая инфекция.
Болезнь Хантингтона возникает в результате дефектного гена (mhTT) на хромосоме номер 4.
Нормальная копия гена вырабатывает белок хантингтин. Дефектный ген больше, чем должен быть. Это приводит к чрезмерному производству цитозина, аденина и гуанина (CAG), строительных блоков ДНК. Обычно CAG повторяется от 10 до 35 раз, но при болезни Хантингтона он повторяется от 36 до 120 раз. Если это повторяется 40 или более раз, симптомы вероятны.
Это изменение приводит к образованию более крупной формы белка хантингтина, который является токсичным. Накапливаясь в головном мозге, он повреждает определенные клетки мозга.
Некоторые клетки мозга чувствительны к более крупной форме хантинтина, особенно те, которые связаны с движением, мышлением и памятью.
Это подрывает их функции и в конечном итоге уничтожает их. Ученые не уверены, как именно это происходит.
Как передается?
Болезнь Хантингтона — аутосомно-доминантное заболевание. Это означает, что он может быть у человека, если он унаследует только одну копию дефектного гена от матери или отца.
У человека с геном есть одна хорошая копия гена и одна неисправная копия.Любое потомство унаследует либо хорошую копию, либо дефектную. У ребенка, унаследовавшего хорошую копию, не разовьется болезнь Хантингтона. Ребенок, унаследовавший ошибочную копию, будет.
У каждого ребенка есть 50% шанс унаследовать дефектный ген. Если они унаследуют дефектный ген, у каждого из их детей будет 50% шанс унаследовать его. Может затронуть несколько поколений.
Человек, не унаследовавший дефектный ген, не заболеет и не сможет передать его своим детям.У ребенка, унаследовавшего дефектный ген, разовьется болезнь Гентингтона, если он достигнет возраста, когда симптомы должны появиться. Около 10% людей с дефектным геном развивают симптомы до 20 лет, а около 10% развивают их после 60 лет.
Если симптомы проявляются в возрасте до 20 лет, это ювенильная болезнь Хантингтона.
Симптомы разные и могут включать скованность в ногах, тремор и регресс в обучении.
Болезнь Хантингтона в настоящее время неизлечима.Лечение не может повернуть вспять или замедлить его прогрессирование.
Однако лекарства и другие методы лечения могут помочь справиться с некоторыми симптомами.
Лекарства
Тетрабеназин (ксеназин) одобрен Управлением по санитарному надзору за качеством пищевых продуктов и медикаментов (FDA) для лечения судорожных, непроизвольных движений или хореи, которые могут возникать при болезни Гентингтона.
Побочные эффекты включают депрессию и суицидальные мысли или действия. Если при приеме этого препарата у человека появляются признаки депрессии или изменения настроения, ему следует немедленно обратиться к врачу.
Третрабеназин не подходит тем, у кого уже есть диагноз депрессии, особенно с суицидальными мыслями.
К лекарствам для контроля движений, вспышек и галлюцинаций могут относиться:
- клоназепам (клонопин)
- галоперидол
- клозапин (клоразил)
Побочные эффекты включают седативный эффект, скованность и ригидность.
При депрессии и некоторых обсессивно-компульсивных расстройствах врач может назначить:
Литий может помочь при сильных эмоциях и изменениях настроения.
Логопедия
Логопедия помогает людям находить способы выражать слова и фразы и более эффективно общаться.
Физиотерапия и трудотерапия
Физиотерапевт может помочь улучшить мышечную силу и гибкость, улучшить равновесие и снизить риск падения.
Эрготерапевт может помочь разработать стратегии управления проблемами концентрации и памяти. Они также могут посоветовать, как сделать дом более безопасным.
Врач осмотрит человека и спросит о его семейном и медицинском анамнезе, а также о симптомах, таких как недавние эмоциональные изменения.
Если они считают, что у человека может быть болезнь Хантингтона, они направят его к неврологу.
Иногда врачи рекомендуют методы визуализации, например компьютерную томографию или МРТ. Они могут выявить изменения в структуре мозга и помочь исключить другие расстройства.
Генетическое тестирование также может помочь подтвердить диагноз.
Болезнь Хантингтона оказывает значительное эмоциональное, психическое, социальное и экономическое влияние на жизнь человека и его близких. Большинство людей с этим заболеванием живут в течение 10–30 лет после постановки диагноза.
Болезнь Гентингтона с ювенильным началом обычно прогрессирует быстрее. Это может привести к летальному исходу в течение 10 лет после постановки диагноза.
Причиной смерти часто бывает осложнение, например пневмония или удушье.
В настоящее время лекарства нет, но лечение может помочь людям справиться с этим заболеванием и улучшить качество их жизни.
Надежда на будущее?
В будущем ученые надеются, что генная терапия найдет решение этой болезни. Исследователи искали способы использования генной терапии для лечения, замедления или предотвращения болезни Хантингтона.
Одна из возможных стратегий — использовать молекулы, известные как синтетические малые интерферирующие РНК (миРНК), для подавления продукции белка из дефектного гена. Это предотвратит накопление токсичного белка Хантингтина и возникновение симптомов.
Однако проблема заключается в том, как доставить миРНК в соответствующие клетки мозга, чтобы они могли быть эффективными.
В 2017 году ученые из Университета Эмори предположили, что методы CRISPR / Cas9, которые включают «вырезание и вставку» ДНК, могут помочь предотвратить болезнь Хантингтона в будущем.
Эксперименты на мышах показали «значительные улучшения» через 3 недели. Большинство следов повреждающего белка исчезли, и нервные клетки показали признаки самовосстановления.
Однако необходимо провести гораздо больше исследований, прежде чем люди смогут это сделать.
Такие организации, как HDSA, предлагают поддержку людям с болезнью Хантингтона и их семьям.
Генетическое тестирование на болезнь Хантингтона стало возможным в 1993 году. Любой, у кого есть семейная история болезни, может спросить своего врача о генетическом тестировании, чтобы выяснить, несут ли они дефектный ген.
Некоторые люди предпочитают выяснять, есть ли у них этот ген и есть ли у них симптомы, в то время как другие предпочитают не знать. В принятии решения может помочь генетический консультант.
Гентингтон, генетика и беременность
Если пара желает иметь ребенка, и у одного из родителей есть дефектный ген, можно пройти курс экстракорпорального оплодотворения (ЭКО).
Затем эмбрион проходит генетическое тестирование в лаборатории и имплантируется женщине только в том случае, если у него нет дефектного гена.
Плод также может пройти генетическое тестирование во время вынашивания, если имеется семейный анамнез заболевания. Это можно сделать с помощью пробы ворсин хориона (CVS) на 10–11 неделе или с помощью амниоцентеза на 14–18 неделе.
Болезнь Хантингтона — Альянс семейных опекунов
Определение
Болезнь Хантингтона (HD), также называемая хореей Хантингтона, является наследственным заболеванием головного мозга, которое приводит к потере физического контроля и умственных способностей. Симптомы обычно появляются в возрасте от 30 до 50 лет, но могут появиться раньше или позже.HD характеризуется прогрессирующим физическим, когнитивным и психологическим ухудшением.
Что происходит, когда у вас HD?
Гены состоят из длинных цепей строительных блоков, называемых нуклеотидами . У человека с HD имеется избыток определенного строительного блока — генетического заикания (участок ДНК, повторяющийся снова и снова на одном конце гена) на четвертой хромосоме. В конечном итоге это приводит к симптомам HD. Поскольку генетическая проблема вызывает потерю нервных клеток в головном мозге, особенно в базальных ганглиях, симптомы HD могут развиваться постепенно и будут влиять на способность человека двигаться, думать и обрабатывать мысли и чувства.Развитие HD часто делится на этапы, и каждый этап знаменует потерю способности или изменение ситуации. По мере прогрессирования БГ в головном мозге повреждаются дополнительные нейроны, что приводит к дальнейшему физическому, когнитивному и психологическому снижению.
Факты
Хотя когда-то считалось редким заболеванием, HD является одним из наиболее часто встречающихся наследственных заболеваний. По всей стране около 30 000 человек страдают болезнью Хантингтона. Поскольку люди, страдающие HD, нуждаются в серьезной заботе со стороны их семей и близких, это разрушительное состояние влияет на широкий круг людей, близких к тем, кто болен.
HD поражает как женщин, так и мужчин, а также все этнические группы. БГ могут развиться у людей от двух до 80 лет. Заболевание постепенно уменьшается в течение 10–25 лет, что обычно приводит к полной зависимости от других. Пострадавший в конечном итоге умирает от таких осложнений, как сердечная недостаточность или аспирационная пневмония.
Примерно 200 000 американцев подвержены риску унаследовать болезнь от больного родителя. Те, у кого есть родитель с HD, имеют 50 процентов (т.е., каждый второй) шанс унаследовать дефектный ген. Другими словами, передача гена HD ребенку случайна; некоторые родители не могут передать ген никому из своих детей; некоторые передают это всем им. HD не пропускает поколения. Следовательно, если ребенок не унаследует ген, он не сможет передать его будущим поколениям. У каждого человека, унаследовавшего ген HD, в конечном итоге разовьется болезнь, если у него не будет других причин (инсульт, рак и т. Д.) До прогрессирования HD.
Симптомы
Человек с болезнью Гентингтона часто имеет проблемы в трех областях: контроль движений тела, когнитивные (или умственные) функции и психологические проблемы.Интенсивность и количество симптомов варьируются от случая к случаю. Симптомы, которые испытывают люди с HD, могут включать:
Движение: Быстрые, танцевальные, неконтролируемые движения конечностей типичны для людей с HD и обычно называются хореей специалистами в области здравоохранения. Наиболее очевидны движения туловища и лица. Все двигательные расстройства имеют тенденцию прогрессировать со временем как по частоте, так и по степени тяжести. Движения могут быть сильно преувеличены, в результате чего то, что задумывалось как небольшое движение, вместо этого станет большим и драматичным.Например, предполагаемый небольшой сдвиг в постели может заставить человека фактически вскочить с постели. Со временем может возникнуть ригидность или жесткость мышц. HD может повлиять на способность вашего любимого человека ходить — он или она может стать неустойчивым или даже казаться «пьяным». На более поздних стадиях заболевания глотание становится все более затруднительным (дисфагия), и серьезное беспокойство вызывает удушье.
Когнитивные: Болезнь Хантингтона вызывает изменения в мозге, которые замедляют обработку информации и препятствуют организационным возможностям.Таким образом, вашему близкому может быть все сложнее организовывать типичные домашние дела, справляться с новыми ситуациями или переключаться с одного задания на другое (многозадачность). Поскольку люди с HD испытывают трудности с обработкой информации, они могут испытывать трудности с запоминанием слов, и речь может становиться затруднительной. Люди с HD могут демонстрировать плохое суждение и проблемы с кратковременной памятью. Снижение когнитивных функций является обычным явлением по мере прогрессирования заболевания, хотя люди с HD продолжают узнавать своих опекунов, знают о том, что происходит вокруг них, и могут давать отзывы о своем состоянии и уходе.
Психологический: Некоторым людям с HD могут быть трудно контролировать импульсы и эмоции, что приводит к вспышкам, крикам или агрессии. От легкой до тяжелой депрессии, часто встречающейся у большинства людей с хроническими длительными состояниями, является одна из основных жалоб на HD. Другие психологические симптомы включают раздражительность и беспокойство. Другие могут демонстрировать шизофреническое поведение, такое как галлюцинации, мания, беспокойство и психоз.
Диагностика болезни Хантингтона
Тщательное неврологическое обследование с использованием унифицированной шкалы оценки болезни Хантингтона (UHDRS) и обширный анамнез пациента и его семьи могут подтвердить наличие HD.Однако проявляющиеся симптомы могут быть вызваны или не быть вызваны заболеванием. Врач или невролог, желательно знакомый с HD, может помочь сделать это определение. Генетическое тестирование — это наиболее убедительный и окончательный способ определить, заболеет ли ваш любимый человек или заболела болезнь Хантингтона.
Ген HD и генетическое тестирование
В 1993 году открытие гена, вызывающего болезнь Хантингтона, привело к разработке прямого генного теста на HD. Этот анализ крови, также называемый пресимптоматическим тестом, может использоваться для прогнозирования болезни Хантингтона до появления симптомов.Тест состоит из серии посещений центра генетического тестирования для генетического консультирования, тщательного неврологического осмотра, психологического интервью, обсуждения положительных или отрицательных результатов и последующего наблюдения. Результаты теста не могут указывать на при появлении симптомов, хотя исследования в этой области продолжаются.
Второй анализ крови, называемый подтверждающим тестом, подтверждает наличие гена HD. Процедура этого теста такая же, как и пресимптоматического теста; однако тест проводится после того, как начали проявляться симптомы заболевания.Доступны пренатальные тесты для определения наличия у плода гена HD. Однако из-за потенциального бремени осведомленности о том, что у человека разовьется это хроническое заболевание, рекомендуется не проходить тестирование до достижения возраста 18 лет. В отсутствие лекарства люди из группы риска могут предпочесть жить в неопределенности, а не проходить тест. Это личное решение, которое следует обсудить с генетическим консультантом, семьями и близкими.
Очень важно найти центр тестирования, который следует рекомендациям Американского общества болезней Хантингтона (HDSA) по генетическому тестированию.HDSA публикует обновленный список этих центров тестирования (см. Ресурсы).
Исследования
Продолжаются исследования по разработке новых стратегий борьбы с HD. В настоящее время исследователи изучают методы отсрочки начала HD или остановки прогрессирования, пока разрабатывается лекарство. Другие исследовательские усилия с моделями на животных определяют эффекты HD, как он проявляется и могут ли успешные испытания на животных быть преобразованы в методы лечения, которые работают на людях. Также ведутся исследования эффектов хирургической трансплантации стволовых клеток и трансплантации тканей.Как базовые (лабораторные), так и клинические (тестирование лекарств и методов лечения) исследования продолжают способствовать тестированию новых наркотиков. Чтобы быть в курсе текущих клинических испытаний или участвовать в них, свяжитесь с вашим местным отделением или национальным офисом HDSA.
Лечение
В настоящее время никакие лекарства не могут остановить или замедлить прогрессирование HD. Таким образом, устранение симптомов является целью лечения HD. Лекарства могут уменьшить непроизвольные движения и эмоциональные расстройства у некоторых пациентов с HD. Нейролептики обычно назначают для уменьшения непроизвольных движений.При психологических симптомах обычно рекомендуются антидепрессанты или антипсихотические препараты. Однако семьям следует проявлять осторожность при использовании новых лекарств, поскольку некоторые люди с HD могут быть более чувствительны к побочным эффектам, чем другие. Важно задокументировать побочные эффекты новых лекарств или изменения в поведении после того, как лекарство было добавлено в распорядок вашего любимого человека.
Многие другие методы лечения могут помочь людям с HD и их близким. Физиотерапевты могут помочь с проблемами равновесия и ходьбы на раннем этапе, а также предложат упражнения для развития силы.Упражнения также помогают бороться с депрессией. Эрготерапевты могут предложить стратегии, которые сделают ваш дом более безопасным и пригодным для жизни для вашего любимого человека с HD, улучшить общение и предложить идеи, чтобы компенсировать снижение когнитивных функций.
Некоторые полезные методы, которые сработали для лиц, осуществляющих уход, включают маркировку предметов в доме (например, для обозначения содержимого кухонного шкафа), использование списков и заметок и общение с помощью коротких прямых инструкций. HD влияет на обмен веществ, и люди, страдающие этим заболеванием, могут сжигать калории намного быстрее, чем в среднем.Таким образом, может быть важно, чтобы ваш любимый человек сидел на высококалорийной диете. Диетологи могут помочь с изменением диеты и планированием питания. Поскольку способность к общению постоянно снижается у людей с HD, дефектолог также может быть особенно полезен на всех стадиях заболевания. В идеале лечение должно быть скоординировано, а лекарства должны назначаться медицинскими работниками, хорошо знакомыми с HD.
Роль медицинских работников
Когда ваш близкий проходит тестирование или получает лечение от HD, вы можете обратиться за помощью к разным специалистам в области здравоохранения.Люди, у которых есть HD, испытывают ряд симптомов и, возможно, много стадий, поэтому вы можете взаимодействовать с тем, что может показаться очень большим количеством врачей, социальных работников, физиотерапевтов и других поставщиков медицинских услуг. Координация ухода и поиск соответствующих ресурсов и специалистов, обученных лечению HD, может оказаться непосильной задачей. Ведение записной книжки или «журнала здоровья», в котором указывается общение с поставщиками медицинских услуг, с которыми вы обращаетесь, назначенное лечение и прописанные лекарства, поможет организовать приемы и обеспечит личный учет ухода.
Обращаясь за ранним вмешательством к специалистам здравоохранения, таким как физиотерапевты, профессиональные терапевты и логопеды, люди с HD могут лучше контролировать болезнь и свое благополучие.
Получение поддержки
Из-за сильного эмоционального воздействия диагноза этого хронического наследственного состояния и стресса от осознания того, что другие члены семьи могут подвергаться риску, участие в группах поддержки может быть очень полезным. Группы поддержки обеспечивают безопасную, заботливую среду, в которой можно делиться информацией об индивидуальном опыте и проблемах HD с другими людьми в аналогичных ситуациях.Обсуждаемые темы могут включать приспособление, выживание, течение болезни, семейные проблемы, разочарования, страхование, лекарства и многое другое. Как онлайн, так и личные группы поддержки доступны для людей с HD, членов их семей, а также для тех, кто живет в группе риска. Консультации также могут быть полезны для отдельных лиц, пар или семей, которые учатся справляться с хроническими заболеваниями или периодическими кризисами со здоровьем.
Семья и друзья, которые оказывают помощь близкому человеку с HD, могут подвергаться повышенному риску плохого здоровья, депрессии и изоляции.Для здоровья члена семьи, обеспечивающего уход, а также здоровья человека с ГБ, важно, чтобы лица, осуществляющие уход за ними, могли отдохнуть от ухода (известная как передышка), высыпаться и иметь собственную систему поддержки. . Временный уход может быть организован с помощью членов семьи, друзей, волонтерских служб, центров самостоятельного проживания, организаций социального обслуживания или агентств по уходу на дому. Воспитатели, которые уделяют время себе, могут лучше заботиться о своих близких. Для получения дополнительной информации см. Информационный бюллетень Family Caregiver Alliance, Забота о ВАС: Самопомощь для семейных опекунов.
Сегодня, более чем когда-либо, есть надежда для людей с HD. В отношении потенциальных методов лечения наблюдается быстрый прогресс. Аналогичным образом, качество ухода за людьми с HD значительно улучшилось за последние годы. Изучая стратегии, помогающие им справиться с HD, люди могут жить полноценной и продуктивной жизнью на последних этапах.
Ресурсы
Альянс семейных опекунов
Национальный центр опеки
(415) 434-3388 | (800) 445-8106
Веб-сайт: www.caregiver.org
Электронная почта: info@caregiver.org
FCA CareNav: https://fca.cacrc.org/login
Государственные услуги: https://www.caregiver.org/connecting-caregivers/services-by-state /
Family Caregiver Alliance (FCA) стремится улучшить качество жизни лиц, осуществляющих уход, посредством образования, услуг, исследований и защиты. Через свой Национальный центр по уходу FCA предлагает информацию по текущим социальным вопросам, государственной политике и вопросам ухода, а также оказывает помощь в разработке государственных и частных программ для лиц, осуществляющих уход.Для жителей большей части района залива Сан-Франциско FCA предоставляет услуги прямой семейной поддержки лицам, ухаживающим за больными Хантингтоном, болезнью Альцгеймера, инсультом, БАС, травмой головы, болезнью Паркинсона и другими изнурительными расстройствами, поражающими взрослых.
Другие организации и ссылки
Huntington’s Disease Society of America
www.hdsa.org
International Huntington Association
www.huntington-assoc.com
HOPES: Охватывающий проект Хантингтона по образованию в Стэнфорде
www.stanford.edu/group/hopes
Этот информационный бюллетень был подготовлен Family Caregiver Alliance и проверен Американским обществом болезней Хантингтона. © 2003 Альянс семейных опекунов. Все права защищены.
Хорея Хантингтона с заболеванием двигательных нейронов | Деменция и когнитивные нарушения | JAMA Neurology
Фон Было несколько сообщений о случаях заболевания двигательных нейронов, связанного с болезнью Хантингтона (HD).
Цель Описать пациента с выраженными фасцикуляциями, хореей и возможным боковым амиотрофическим склерозом (БАС), у которого генетическое тестирование выявило мутацию HD.
Дизайн История болезни.
Настройка Юго-западный медицинский центр Техасского университета, Даллас.
Пациент 69-летний мужчина с хореей и фасцикуляциями.
Вмешательства Генетическое и электрофизиологическое тестирование.
Основные показатели результатов Результат генетического теста, результат электрофизиологического теста и физикального обследования.
Результаты У 69-летнего мужчины с давней депрессией и нарушением памяти были подергивания мышц продолжительностью 8 месяцев. При неврологическом обследовании у него были обнаружены хореоатетоидные движения и дистальная слабость. Электрофизиологические исследования выявили признаки заболевания двигательных нейронов. Генетический тест показал повторение 40 CAG на хромосоме 4, что подтвердило диагноз HD.
Заключение Заболевание двигательных нейронов может редко возникать у пациентов с HD и может быть одной из его характерных черт.
Болезнь Хантингтона (БХ) — аутосомно-доминантное нейродегенеративное расстройство, характеризующееся когнитивной дисфункцией, прогрессирующими поведенческими аномалиями и аномальными движениями, включая хорею, атетоз, ригидность или дискинезию. Это вызвано экспансией тринуклеотидного (CAG) повтора в гене хантингтина, расположенном на коротком плече хромосомы 4. 1 Симптомы и признаки, относящиеся к дисфункции двигательных нейронов, обычно не признаются как часть процесса HD.
Мы описываем пациента, который сообщил о мышечных подергиваниях, у которого фасцикуляции и хореоатетоидные движения были очевидны во всех конечностях при первоначальном обследовании. Электромиография выявила признаки диффузной активной денервации и фасцикуляций. Последующие результаты генетического тестирования показали увеличение количества повторов CAG в гене хантингтина.
69-летний правша с историей депрессии в течение 9 лет поступил в нервно-мышечную клинику Юго-западного медицинского центра Техасского университета в Далласе для оценки мышечных сокращений всех конечностей в течение 8 месяцев. Случайных перемещений не было. Однако ему пришлось уйти с должности адвоката из-за проблем с памятью, отмеченных за 2 года до выступления. Изменений личности не отмечено.Предыдущее лечение включало мемантин и различные антидепрессанты при расстройствах настроения и когнитивных расстройствах. У него не было бульбарных симптомов, одышки, парестезий или недержания мочи. Его история болезни имела большое значение для гипертонии и гиперлипидемии. Семейный анамнез не имел никакого отношения. Его отец и мать умерли в возрасте 93 и 97 лет соответственно. У него было 2 брата. Один брат умер от лейкемии; другой умер от новообразования головного мозга. В анамнезе не было никаких известных побочных движений или расстройства двигательных нейронов у членов основной и расширенной семьи.
При медицинском осмотре он был внимателен и ориентирован. У него были непрерывные беспорядочные нервные движения всеми частями тела, включая сгибание пальцев, поджимание рта, поднятие лба, перекатывание бедер и метание головы. Гинекомастии у него не было. Черепные нервы в норме. Рывок челюстью отсутствовал. Грубые фасцикуляции присутствовали в руках, бедрах, икрах и верхней части груди. Мышечный тонус по всему телу был нормальным. Внутренние мышцы кисти атрофированы. Наблюдалась слабость от легкой до умеренной во внутренних мышцах кисти, тыльных сгибателях голеностопного сустава и разгибателях пальцев ног с обеих сторон.Сохранены глубокие сухожильные рефлексы. Сенсорное исследование не повлияло на модальности мелких и крупных волокон. Координация нормальная. Походка отличалась легким двусторонним отвисанием стопы.
Следующие результаты анализов крови были нормальными или отрицательными: гемограмма, морфология эритроцитов, общая железосвязывающая способность, комплексная метаболическая панель, скорость оседания эритроцитов, электрофорез сывороточного белка, активность гексозаминидазы лейкоцитов и уровни витамина B 12 , фолиевая кислота, тиамин, витамин Е, медь, тиреотропин и паратироидный гормон.Результаты серологического тестирования на сифилис, вирус иммунодефицита человека, болезнь Лайма и человеческий Т-лимфотрофный вирус 1 были отрицательными. Антитела к тиреоглобулину, ганглиозидозу GM1, ганглиозиду GD1B и тканевой трансглутаминазе IgA отсутствовали; панель паранеопластических антител была отрицательной (включая цитоплазматические антитела клеток Пуркинье типа 1 и антитела к белку 5, опосредованные ответом на коллапсин). Титр сывороточных антинуклеарных антител составлял 1: 160 (референсный диапазон <1: 160). Креатинкиназа сыворотки была повышена до 563 (референсный диапазон, 40-210 Ед / л) (для преобразования в микрокаталы на литр, умножьте на 0.0167), а уровень церулоплазмина в сыворотке был 24,4 (референсный диапазон 14,0–21,9 мг / дл).
Результаты исследований нервной проводимости показали снижение амплитуды потенциалов действия сложных мышц в правом срединном (2,3 мВ; референтный диапазон> 4 мВ) и малоберцовых (0,7 мВ; референтный диапазон> 2 мВ) нервах. Правые локтевые и большеберцовые двигательные и сенсорные реакции были нормальными. Игольчатая электромиография показала диффузную умеренно активную денервацию и фасцикуляции шейных, грудных и пояснично-крестцовых миотомов.
Магнитно-резонансная томография головного мозга с контрастной средой показала умеренные изменения сигнала перивентрикулярного белого вещества, соответствующие микрососудистой ишемии. Магнитно-резонансная томография шейного отдела позвоночника выявила 3-мм антеролистез С5 относительно С6 с гиперинтенсивным изменением сигнала спинного мозга на уровне С5. Нейропсихиатрическая оценка показала слабое или умеренное нарушение памяти, при этом вербальная память затрагивала больше, чем зрительную. Управляющая функция и внимание остались нетронутыми.Генетическое тестирование показало 40 и 18 повторов CAG для каждого из 2 аллелей гена хантингтина IT15 , что подтверждает диагноз HD.
Учитывая, что шейная спондилотическая миелопатия потенциально излечима и могла способствовать его слабости, ему была сделана операция по поводу декомпрессии спинного мозга. Однако в течение следующих нескольких месяцев слабость значительно усилилась с дальнейшим прогрессированием в проксимальных группах мышц верхних и нижних конечностей. У него также развилась дисфагия.В его слабых мышцах сохранились глубокие сухожильные рефлексы со знаком скрещенной приводящей мышцы слева. Похоже, что его хорея и когнитивная дисфункция не ухудшились в течение этого периода.
Связь хореи с заболеванием двигательных нейронов встречается редко и может наблюдаться при различных неврологических расстройствах, таких как болезнь Вильсона, системная красная волчанка, эндокринопатии, нейроакантоцитоз, ганглиозидоз GM2, заболевание тела полиглюкозана у взрослых, дентаторубралпаллидолуизиальная атрофия белковой атрофии.Восемь пациентов с одновременным боковым амиотрофическим склерозом (БАС) и хореей были описаны до 1993 года, когда впервые стало доступно генетическое тестирование на HD. 2 Rubio et al. 2 впоследствии описали первого пациента с генетически подтвержденным HD, у которого после более чем 10 лет типичных симптомов HD развилось сочетание признаков верхних и нижних мотонейронов с явными патологическими признаками БАС. 2 При вскрытии типичные гистологические данные HD были очевидны в полосатом теле и других частях мозга.В спинном мозге наблюдалась серьезная потеря клеток переднего рога, тельца бунина и клубковидные включения. Выжившие клетки переднего рога продемонстрировали иммунореактивность к супероксиддисмутазе 1. Семейный анамнез у этой пациентки был значимым для материнского БАС без побочных движений. Было неясно, представляло ли это возникновение 2 отдельных болезненных процессов или результат прогрессирования патологии HD на двигательные нейроны. В последние годы были описаны 3 пациента с одновременным генетически подтвержденным HD и ALS. 1 , 3 , 4 Постулируется несколько возможных механизмов потери двигательных нейронов при HD. Неправильная укладка белков, транскрипция ДНК, вмешательство в процессинг РНК, стимулирование апоптоза и дисфункция цитоплазматических элементов 5 , а также эксайтотоксичность глутамата 6 вовлечены в токсический процесс, ведущий к потере нервных клеток.
Также была описана хорея у пациента с БАС без HD. Патофизиология побочных движений у этих пациентов остается неясной.Pradat et al. 7 описали 40-летнего мужчину, у которого развились прогрессивные хореические движения через 6 лет после постановки диагноза клинически определенного БАС. 7 Результаты обширного тестирования причин хореи, включая генетическое тестирование на HD, спиноцеребеллярную атаксию типов 1, 2, 3, 6 и 7, а также на дентаторноубралпаллидолизианскую атрофию, были отрицательными. Было высказано предположение, что длительное течение БАС у их пациента привело к дегенерации экстрапирамидных структур с последующим развитием гиперкинетических движений.Однако развитие гиперкинетических движений остается спекулятивным, поскольку о патологических исследованиях не сообщалось. Среди начальных патологических исследований Ludolph и Knirsch 8 сообщили о результатах вскрытия у пациента с подозрением на БАС, у которого развились гемибаллизм и хореоатетоз через 2 с половиной года после начала БАС. Тестирование генов болезни Хантингтона не проводилось. Пациент умер через 2 недели после появления экстрапирамидных симптомов. Вскрытие показало потерю мотонейронов в черной субстанции, бледном мозге и надъядерных центрах движений глаз.Иммуногистохимическое исследование выявило внутриядерные убиквитин-положительные включения в двигательных нейронах. В другом патологическом отчете пациента с семейным БАС, у которого была хорея и баллизм, патологоанатомическое исследование выявило заметную потерю нейронов и глиоз в подталамусе, внутреннем бледном шаре, компактной части черной субстанции и красном ядре. Интересно, что хвостатое тело, скорлупа и таламус были нормальными. В мотонейронах обнаружены редкие убиквитин-положительные клубковидные включения. Результаты генетических тестов на HD, супероксиддисмутазу 1 и дентаторубралпаллидолизианскую атрофию были отрицательными. 9
В литературе описаны другие формы прогрессирующего расстройства нижних мотонейронов в сочетании с экстрапирамидными симптомами. 10 -12 О некоторых из этих ассоциаций сообщалось до эры тестирования генов HD. 11 , 12
В отличие от предыдущих статей, в отчете нашего пациента были мышечные судороги продолжительностью 8 месяцев. Первоначальное неврологическое обследование выявило обильные фасцикуляции и небольшую дистальную слабость, а также хореоатетоидные движения.Временное совпадение вовлечения двигательных нейронов и полосатого тела у нашего пациента с мутацией гена HD свидетельствует о том, что ранняя HD проявляется в обеих нейронных системах, а не в случайном возникновении независимых болезненных процессов. В течение следующих нескольких месяцев его слабость быстро прогрессировала и затронула несколько других групп мышц, как и ожидается при БАС. Его хорея оставалась стабильной и не приводила к инвалидности. Представление было осложнено шейной спондилотической миелопатией. Однако продолжающееся прогрессирование слабости конечностей после хирургической коррекции свидетельствует против ее влияния на симптомы пациента.Таким образом, мы предполагаем, что заболевание двигательных нейронов может быть одной из характерных черт HD.
Для корреспонденции: Джая Р. Триведи, доктор медицины, отделение неврологии, Юго-западный медицинский центр Техасского университета, 5323 Harry Hines Blvd, Даллас, Техас 75390-8897 (jaya.thibitedi@utsouthwestern.edu).
Принята к публикации: 29 июня 2010 г.
Вклад авторов: Концепция и дизайн исследования : Садегиан, Баттисте и Триведи. Сбор данных : Садегиан, О’Суйлеабхайн, Эллиотт и Триведи. Анализ и интерпретация данных : Садегиан, Эллиотт и Триведи. Составление рукописи : Садегян. Критический пересмотр рукописи на предмет важного интеллектуального содержания : Садегиан, О’Суйлеабхайн, Баттист и Триведи. Административная, техническая и материальная поддержка : Садегиан, О’Суйлеабхайн, Эллиотт и Триведи. Кураторская работа : Триведи.
Раскрытие финансовой информации: Д-р O’Suilleabhain получил исследовательский грант от NIH, Neurosearch и Cure Huntington Disease Initiative для болезни Хантингтона, Impax Pharmaceuticals и по делам ветеранов по болезни Паркинсона и Allergan по борьбе с дистонией. Он также получал гонорары за консультации или выступление от Teva Pharmaceuticals, Allergan и Ipsen Group за последние 2 года.
1.Канаи Ккувабара SSawai S и другие.Генетически подтвержденная болезнь Хантингтона, маскирующаяся под болезнь двигательных нейронов. Mov Disord 2008; 23 (5) 748-751PubMedGoogle ScholarCrossref 2.Rubio ASteinberg К.Фиглевич DA и другие. Сосуществование болезни Хантингтона и семейного бокового амиотрофического склероза: описание случая. Acta Neuropathol 1996; 92 (4) 421- 427PubMedGoogle ScholarCrossref 3.Papageorgiou С.Г.Антелли ABonakis А и другие.Связь генетически доказанной болезни Хантингтона и спорадического бокового амиотрофического склероза у 72-летней женщины. J Neurol 2006; 253 (12) 1649–1650 PubMedGoogle ScholarCrossref 4. Пхукан Джали EPender НП и другие. Болезнь Хантингтона, проявляющаяся в виде бокового амиотрофического склероза. Боковой склер амиотрофа 2010; 11 (4) 405-407PubMedGoogle ScholarCrossref 6. Розенберг Р. Н. Амиотрофия при мультисистемных генетических заболеваниях.Роуленд LP Advances in Neurology New York, NY Raven Press 1982; 149-158 Болезни двигательных нейронов человекаvol 36Google Scholar7.Pradat PFSalachas FLacomblez L и другие. Ассоциация хореи и болезни двигательных нейронов. Mov Disord 2002; 17 (2) 419-420PubMedGoogle ScholarCrossref 9.Gamez JCorbera-Bellalta MMila MLópez-Lisbona РБолуда SFerrer I Хорея-баллизм, связанный с семейным боковым амиотрофическим склерозом: клиническое, генетическое и невропатологическое исследование. Mov Disord 2008; 23 (3) 434- 438PubMedGoogle ScholarCrossref 10.Pageot NVial CRemy CChazot GBroussolle E Прогрессирующая хорея и амиотрофия без акантоцитов: новый случай синдрома Фотопулоса? Дж Нейрол 2000; 247 (5) 392-394PubMedGoogle ScholarCrossref 11.Fotopulos D. Хорея Хантингтона и хроническая прогрессирующая спинальная мышечная атрофия. Psychiatr Neurol Med Psychol (Лейпц) 1966; 18 (2) 63-70PubMedGoogle Scholar12.Серратрис GCremieux GPellissier JFPouget J 2 случая синдрома Фотопулоса (хроническая спинномозговая амиотрофия плечевого пояса и хроническая хорея) [на французском языке]. Пресс Мед 1984; 13 (20) 1274PubMedGoogle ScholarБолезнь Хантингтона | Определение, причина, симптомы и лечение
Болезнь Хантингтона , также называемая Хорея Хантингтона , относительно редкое и неизменно фатальное наследственное неврологическое заболевание, которое характеризуется нерегулярными и непроизвольными движениями мышц и прогрессирующей потерей когнитивных способностей. .Заболевание впервые было описано американским врачом Джорджем Хантингтоном в 1872 году.
Британская викторина
Болезни, расстройства и многое другое: медицинская викторина
Какое состояние вызвано отложением солей мочевой кислоты? Как еще называют перелом костей? Узнайте, что вы знаете о болезнях, расстройствах и многом другом.
Симптомы болезни Хантингтона обычно проявляются в возрасте от 35 до 50 лет и со временем ухудшаются. Они начинаются с случайных подергиваний или извивающихся движений, называемых хореиформными движениями, или с того, что кажется незначительными проблемами с координацией; эти движения, которые отсутствуют во время сна, ухудшаются в течение следующих нескольких лет и переходят в случайные, неконтролируемые и часто сильные подергивания и подергивания. Могут появиться симптомы ухудшения психического состояния, включая апатию, усталость, раздражительность, беспокойство или капризность; эти симптомы могут прогрессировать до потери памяти, слабоумия, биполярного расстройства или шизофрении.
Ребенок человека с болезнью Гентингтона имеет 50-процентный шанс унаследовать генетическую мутацию, связанную с этим заболеванием, и у всех людей, унаследовавших эту мутацию, в конечном итоге разовьется болезнь. Генетическая мутация, вызывающая болезнь Хантингтона, происходит в гене, известном как HD (официально называемый гентингтином [болезнь Хантингтона]). Этот ген, расположенный на хромосоме 4 человека, кодирует белок под названием хантингтин, который распределяется в определенных областях мозга, а также в других тканях тела.Мутировавшие формы гена HD содержат аномально повторяющиеся сегменты дезоксирибонуклеиновой кислоты (ДНК), называемые тринуклеотидными повторами CAG. Эти повторяющиеся сегменты приводят к синтезу белков хантингтина, которые содержат длинные участки молекул аминокислоты глутамина. Когда эти аномальные белки хантингтина разрезаются на фрагменты во время обработки клеточными ферментами, молекулы глутамина выступают из концов фрагментов белка, заставляя фрагменты прикрепляться к другим белкам.Образовавшиеся скопления белков могут вызвать дисфункцию нейронов (нервных клеток). Образование аномальных белков хантингтина приводит к дегенерации и, в конечном итоге, гибели нейронов в базальных ганглиях, паре нервных кластеров в глубине мозга, которые контролируют движение.
Прогрессирование и тяжесть болезни Хантингтона связаны с длиной области тринуклеотидного повтора CAG в гене HD . Например, тринуклеотидная область в HD , по-видимому, расширяется в среднем возрасте, совпадая с началом симптомов, а также может расширяться от одного поколения к другому, вызывая форму болезни, известную как ожидание, при которой симптомы развиваются в более ранний возраст потомков пораженных особей.
Получите подписку Britannica Premium и получите доступ к эксклюзивному контенту. Подпишитесь сейчасНесмотря на то, что существует генетический тест на болезнь Хантингтона, нет эффективных методов лечения или лечения этого расстройства, хотя хореиформные движения могут быть частично и временно подавлены фенотиазинами или другими антипсихотическими препаратами.
Болезнь Гентингтона и хорея — одно и то же?
Болезнь Хантингтона — редкое генетическое (унаследованное от родителей) заболевание, поражающее мозг.По оценкам, от трех до семи человек из 100 000 живут с болезнью Хантингтона. А на запущенных стадиях человек может умереть от осложнений. Хотя лекарства нет, его поиску посвящается все больше исследований. Существуют также методы лечения, которые могут дать надежду и улучшить качество жизни человека.
Почему это называется болезнью Хантингтона?
В конце 1800-х годов доктор Джордж Хантингтон был одним из первых врачей, наблюдавших и изучавших эту болезнь.Он опубликовал знаменитую статью «О хореи», в которой подробно описал семьи Ист-Хэмптона, которые страдали от этого необычного состояния. Со временем она стала известна как «хорея Хантингтона», но поскольку не все, кто страдает этим заболеванием, страдают хореей, название было изменено на «болезнь Хантингтона».
Тогда что такое хорея?
Хорея, которая иногда является симптомом болезни Хантингтона, но не смертельным, является одним из нескольких известных непроизвольных движений, которые также включают более распространенные, такие как тремор и тики.Для неподготовленного глаза может быть сложно определить хорею, потому что ее внешний вид варьируется от одного человека к другому. Причины включают плохую реакцию на лекарства, используемые для лечения поведенческих и желудочно-кишечных расстройств, ревматическую лихорадку, беременность и болезнь Хантингтона.
Каковы признаки болезни Гентингтона?
В 90 процентах случаев симптомы появляются в возрасте от 30 до 50 лет. На самой ранней стадии у человека могут быть проблемы с мышлением или изменение настроения, проблемы на работе и напряженные отношения с другими людьми.На этом этапе важно посоветовать членам семьи, что симптомы болезни Хантингтона часто включают нечто большее, чем просто хорею. Фактически, хорея может появиться только через много лет. Именно тогда появляются другие моторные (двигательные) симптомы, чаще всего в руках, ногах и лице, вызывающие неуравновешенность и неуклюжесть. На поздних стадиях болезни Хантингтона человеку трудно выполнять повседневные действия, такие как ходьба, разговор и глотание.
Как диагностируется болезнь Хантингтона?
Установить диагноз не всегда просто.Ранние психологические симптомы неспецифичны для болезни Хантингтона. Они могут быть признаками другого заболевания, такого как психоз, алкоголизм или биполярное расстройство, или вовсе отсутствовать заболевание. Учитывая редкость болезни Гентингтона, признаки хореи и других аномальных движений, которые являются относительно легкими, могут не распознаваться неподготовленным глазом. В Stony Brook мы проводим тщательное неврологическое обследование, интервьюируем человека и изучаем его семейный анамнез. Если все еще есть сомнения, доступен генетический тест.
Каковы плюсы и минусы генетического тестирования на болезнь Хантингтона?
Генетический тест может подтвердить наличие или отсутствие аномалии гена ДНК, которая вызывает болезнь Хантингтона. Однако пройти генетическое тестирование не всегда полезно. Это потому, что не все люди с аномалией гена Хантингтона болеют этим заболеванием. А некоторые не доживают до появления симптомов. Для других лекарство может стать доступным задолго до начала болезни.Окончательный диагноз может быть поставлен человеку с классическими симптомами и неврологическими признаками, семейным анамнезом и наличием аномалии гена Хантингтона.
Какие существуют виды лечения?
Хотя не существует лекарства или лечения, которое замедляет прогрессирование болезни Хантингтона или обращает ее вспять, существуют методы лечения, которые могут улучшить качество жизни. К ним относятся терапия и лекарства от симптомов настроения, лекарства от хореи, а также физиотерапия и профессиональная терапия, которые помогают человеку выполнять повседневные действия.
Что отличает Стоуни-Брук?
Наша больница недавно вошла в 4% лучших по неврологии и нейрохирургии в стране. У нас есть один из четырех центров передового опыта, назначенных Американским обществом болезней Хантингтона в штате Нью-Йорк. (Их всего 50 по всей стране.) В нашу команду входят специалисты в области неврологии, психиатрии, психологии, нейропсихологии, физиотерапии и патологии речи. Мы консультируемся со специалистами в области медицинской генетики, репродуктологии и акушерства, трудотерапии и питания.Наши пациенты также имеют доступ к клиническим испытаниям и виртуальным встречам групп поддержки.
За дополнительной информацией обращайтесь к Эрин Харабес, LMHC, по телефону (631) 444-3448. Чтобы записаться на прием к одному из наших специалистов по болезни Хантингтона, позвоните Изабель Мелендес: (631) 444-2599.
stonybrookmedicine.edu/huntingtonsdisease